
記住我
Osteoarthritis (OA) is a highly prevalent and destructive degenerative joint disease, affecting approximately 7.6% of the global population, with over 595 million cases in 2020 alone (GBD, 2021 Osteoarthritis Collaborators, 2023). Its incidence is expected to approach 1 billion by 2050, driven by the aging population (GBD, 2021 Osteoarthritis Collaborators, 2023). This rising prevalence poses a significant public health challenge (Martel-Pelletier et al., 2016; Grandi and Bhutani, 2020). The pathological features of OA include cartilage degeneration, synovial inflammation, subchondral bone remodeling, and joint space narrowing, among others (Barnett, 2018; Zhou et al., 2024). Traditionally, OA has been attributed primarily to excessive mechanical load; however, recent research indicates that it is a multifactorial process involving inflammation, metabolic imbalance, and impacts on various joint structures (Barnett, 2018; Hunter and Bierma-Zeinstra, 2019; Zhou et al., 2024). Despite extensive studies revealing some molecular mechanisms and risk factors associated with OA, the exact pathogenesis remains unclear, and effective treatments to halt or slow disease progression are lacking (Henrotin, 2022; Salman et al., 2023). Clinically, OA management predominantly focuses on symptom relief and surgical intervention, underscoring the urgent need to explore its underlying mechanisms and identify new therapeutic targets (Hunter et al., 2020; Kolasinski et al., 2020; Cho et al., 2021; Loughlin, 2022; Mobasheri et al., 2023).
With advancements in the study of cell death mechanisms, ferroptosis—a form of cell death distinctly different from traditional apoptosis and necrosis—has been identified (Dixon and Pratt, 2023; Wahida and Conrad, 2023; Berndt et al., 2024). Ferroptosis involves iron-dependent lipid peroxidation, leading to disrupted iron homeostasis, excess ROS production, and increased lipid peroxidation byproducts (Chen et al., 2023; Deng et al., 2023; von Krusenstiern et al., 2023; Bell et al., 2024; Dixon and Olzmann, 2024). Ferroptosis, discovered in 2012, is now recognized as a key factor in various diseases, including neurodegenerative disorders, cancer, and ischemia-reperfusion injury (Dixon et al., 2012; Deng et al., 2023; Fang et al., 2023; Graham et al., 2023; Miao et al., 2023; Yao et al., 2023; Ahola and Langer, 2024; Levi et al., 2024). Initial research indicates that ferroptosis could be a primary mechanism responsible for OA’s death of chondrocytes and synoviocytes (Miao et al., 2022; He Q et al., 2023). Increased intra-articular iron levels, lipid peroxidation, and weakened antioxidant defenses promote ferroptosis, worsening cartilage degeneration, and joint function loss (Chen J et al., 2024). This finding provides fresh insight into the pathogenesis of OA and presents potential for developing new therapeutic approaches aimed at targeting ferroptosis.
Although research on the precise mechanisms and clinical relevance of ferroptosis in OA is still in its infancy, the volume of related studies is growing swiftly (Yang et al., 2021; Ohnishi et al., 2022; Wang et al., 2023). A thorough review of the molecular pathological mechanisms, potential physiological functions, and therapeutic possibilities of ferroptosis in OA is thus essential. This review systematically outlines the core mechanisms of ferroptosis and its specific impacts on OA, explores the potential of ferroptosis-related biomarkers as diagnostic and therapeutic targets, and discusses the challenges and opportunities for clinical translation.
2 Biological mechanisms of ferroptosis2.1 Overview of ferroptosisFerroptosis is a distinct type of programmed cell death, setting itself apart from conventional forms like apoptosis, necrosis, and autophagy. Its uniqueness lies in its dependence on iron accumulation, which triggers lipid peroxidation (Dixon et al., 2012; Chen et al., 2016; Zhang et al., 2018; Frank and Vince, 2019; Stockwell and Jiang, 2020; Liu et al., 2022; Tang et al., 2024). Morphologically, ferroptosis is characterized by alterations in mitochondrial structure, such as increased mitochondrial membrane density, diminished or missing cristae, and overall mitochondrial shrinkage (Wang et al., 2017; Fang et al., 2019; Deng et al., 2023; Mao et al., 2023). Biochemically, ferroptosis is indicated by the inhibition of cystine/glutamate antiporter (System Xc−), the depletion of glutathione (GSH), decreased activity of glutathione peroxidase 4 (GPX4), accumulation of lipid peroxides (LOOH), and a decline in mitochondrial membrane potential (Mao et al., 2021; Wang and Min, 2021). These changes result in excessive intracellular iron accumulation, triggering oxidative stress and cellular damage (Ingold et al., 2018; Bersuker et al., 2019; Zheng and Conrad, 2020).
Current research on the mechanisms of ferroptosis focuses on three primary signaling axes: dysregulated iron metabolism, lipid peroxidation, and the System Xc−-GSH-GPX4 axis. Dysfunctions in these pathways can lead to cellular iron overload. To maintain iron homeostasis, cells employ intricate regulatory mechanisms, including iron uptake via the transferrin receptor (TFR1), storage of iron in ferritin, and ferritinophagy to mitigate the toxic effects of iron overload (Kakhlon and Cabantchik, 2002; Gammella et al., 2015; Anderson and Frazer, 2017; Feng et al., 2020). GPX4 plays a critical role in counteracting ferroptosis by mitigating lipid peroxidation, while other factors like nicotinamide adenine dinucleotide phosphate (NADPH) and coenzyme Q10 (CoQ10) also contribute to ferroptosis regulation (Bersuker et al., 2019).
In addition to these well-characterized mechanisms, recent studies have highlighted alternative ferroptosis surveillance pathways that operate independently of GPX4. Enzymes such as FSP1 and MBOAT1/2 have been identified as important contributors to these pathways, suggesting a broader and more complex regulatory network of ferroptosis beyond the conventional System Xc−-GSH-GPX4 axis (Liang et al., 2023). The complex biological network governing ferroptosis highlights its central role in cellular homeostasis and disease progression. This understanding provides a crucial foundation for exploring ferroptosis-driven mechanisms in osteoarthritis. Figure 1 offers a comprehensive depiction of the critical molecular mechanisms underpinning ferroptosis.
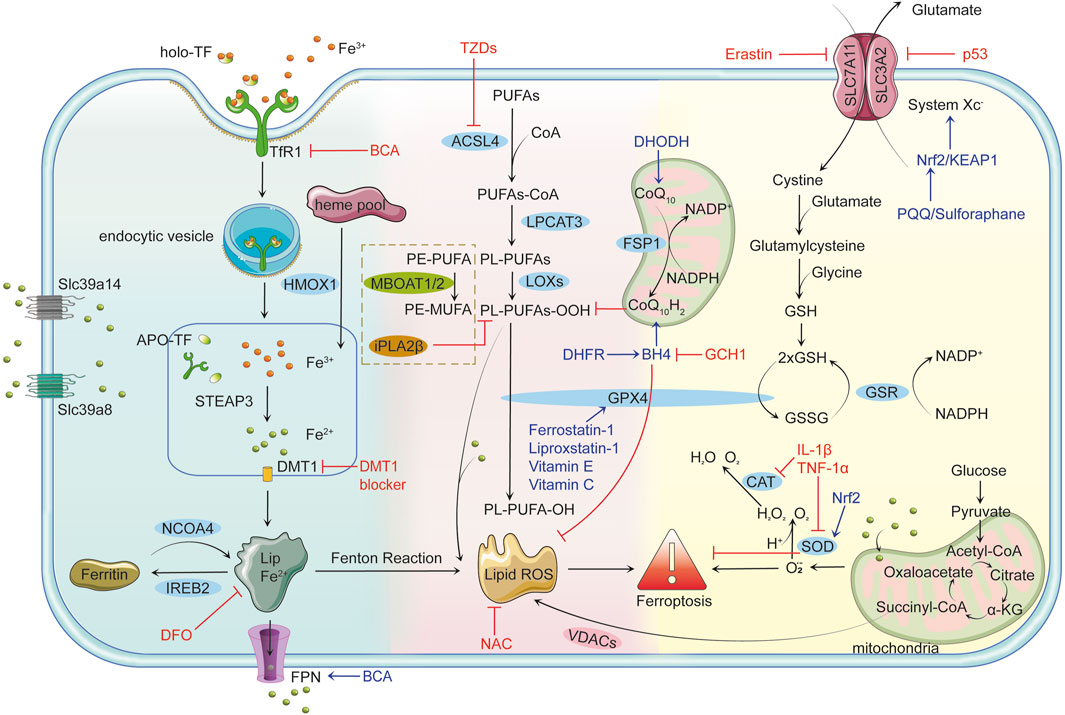
Figure 1. Mechanisms of Ferroptosis Iron Metabolism and Ferroptosis: Cells uptake iron-bound transferrin (TF) through transferrin receptor 1 (TFR1). Iron enters the cell in the Fe³⁺ form via endocytosis and is reduced to Fe2⁺ by six-transmembrane epithelial antigen of prostate 3 (STEAP3) in the endosome, subsequently transported to the cytoplasm by divalent metal transporter 1 (DMT1). Non-transferrin-bound iron (NTBI) uptake, mediated by solute carrier family 39 member 14 (Slc39a14) and solute carrier family 39 member 8 (Slc39a8), further contributes to intracellular iron overload. Free Fe2⁺ can participate in the Fenton reaction, producing highly reactive hydroxyl radicals (•OH) that trigger lipid peroxidation and lead to the accumulation of lipid reactive oxygen species (Lipid ROS). Ferritin alleviates iron toxicity by storing iron, while Nuclear Receptor Coactivator 4 (NCOA4) mediates ferritin degradation, releasing iron ions and enhancing ferroptosis. Lipid Peroxidation and Ferroptosis: Polyunsaturated fatty acids (PUFAs) are catalyzed by long-chain fatty acid-CoA ligase 4 (ACSL4) to form PUFAs-CoA, which are then integrated into the lipid bilayer by lysophosphatidylcholine acyltransferase 3 (LPCAT3), producing phospholipid PUFAs (PL-PUFAs). Lipoxygenases (LOXs) further oxidize PL-PUFAs into phospholipid peroxides (PL-PUFA-OOH), and the accumulated lipid ROS triggers ferroptosis. Voltage-dependent anion channels (VDACs) facilitate the accumulation of lipid ROS, accelerating cell death. Glutathione peroxidase 4 (GPX4)-independent pathways, such as iPLA2β (a calcium-independent phospholipase A2 family member) and membrane-bound O-acyltransferase 1/2 (MBOAT1/2), provide additional defense against ferroptosis by reducing peroxidized lipids and remodeling phospholipids, respectively. Antioxidant Defense Mechanisms and Ferroptosis: GPX4 is the primary inhibitor of ferroptosis, reducing PL-PUFA-OOH to non-toxic phospholipids (PL-PUFA-OH) using reduced glutathione (GSH) to prevent lipid ROS accumulation. GSH synthesis depends on System Xc−, which consists of solute carrier family seven member 11 (SLC7A11) and solute carrier family three member 2 (SLC3A2) and facilitates the transmembrane exchange of cystine and glutamate. Inhibition of System Xc− decreases GSH production, leading to lipid peroxidation and ferroptosis. Key Regulatory Factors and Drug Actions: p53 limits GSH synthesis by inhibiting System Xc−, promoting ferroptosis. The nuclear factor erythroid 2-related factor 2 (Nrf2)/kelch-like ech-associated protein 1 (KEAP1) pathway enhances cellular antioxidant capacity and inhibits ferroptosis. Tetrahydrobiopterin (BH4), synthesized by gtp cyclohydrolase 1 (GCH1), boosts antioxidant defenses and suppresses lipid peroxidation. Ferroptosis suppressor protein 1 (FSP1) participates in antioxidant reactions through coenzyme Q10 (CoQ10), inhibiting ferroptosis. Erastin promotes ferroptosis by suppressing System Xc− and reducing GSH levels. Ferrostatin-1 and Liproxstatin-1 prevent ferroptosis by inhibiting lipid peroxidation, while deferoxamine (DFO) chelates free iron to prevent ferroptosis by blocking the Fenton reaction. N-acetylcysteine (NAC) enhances antioxidant capacity by increasing GSH synthesis. Thiazolidinediones (TZDs) reduce lipid peroxide formation by inhibiting ACSL4 activity. Connection between Ferroptosis and Inflammatory Responses: Pro-inflammatory factors such as interleukin-1β (IL-1β) and tumor necrosis factor-alpha (TNF-α) can induce ROS production, facilitating ferroptosis. Concurrently, ferroptosis can activate inflammatory responses, exacerbating cellular damage. Role of Mitochondria in Ferroptosis: Mitochondria produce energy via oxidative phosphorylation and the tricarboxylic acid (TCA) cycle while generating superoxide (O₂•⁻), which is converted to H₂O₂ by superoxide dismutase (SOD). H₂O₂ reacts with Fe2⁺ to form OH•, further aggravating lipid peroxidation. Additionally, mitochondria release ROS through VDACs, closely linking this process to ferroptosis. Dihydroorotate dehydrogenase (DHODH) regulates ferroptosis by influencing mitochondrial redox reactions.
2.1.1 Iron metabolismIron metabolism and regulation are crucial for maintaining normal cellular functions (Ward et al., 2014; Zheng and Conrad, 2020; Simao and Cancela, 2021). Transferrin receptor 1 (TfR1) mediates the endocytosis of iron-bound transferrin (TF) (Aisen et al., 1978; Johnsen et al., 2019). TF, a natural chelator abundant in plasma, possesses two high-affinity iron-binding sites. Once TF binds to Fe³⁺ to form holo-TF, it associates with TfR1 on the cell surface and is internalized. In the acidic environment of endosomes, the lowered pH facilitates the release of Fe³⁺ from the Tf-TfR1 complex, allowing TfR1 and apotransferrin (apoTF) to be recycled back to the cell surface (Klausner et al., 1983; Zhou et al., 2021). During this process, divalent metal transporter 1 (DMT1) is crucial for the transmembrane transport of iron, primarily responsible for transporting iron ions from the extracellular space or organelles into the cytoplasm. Metalloreductase six-transmembrane epithelial antigen of prostate 3 (STEAP3) possesses ferrireductase activity, reducing Fe³⁺ to Fe2⁺, which is then released into the cytoplasm via DMT1, contributing to an unstable iron pool (Pantopoulos, 2004; Feng et al., 2020; Koike et al., 2020). Excess iron is stored in ferritin or exported extracellularly through ferroportin 1 (FPN1) (Kakhlon and Cabantchik, 2002).
Intracellular iron overload is a key driver of ferroptosis, facilitated by increased iron uptake and ferritin degradation through ferritinophagy. In ferroptosis-sensitive cells, TfR1 expression is upregulated, enhancing iron uptake, while ferritin heavy chain (FTH1) and ferritin light chain (FTL) undergo selective degradation via ferritinophagy (Santana-Codina and Mancias, 2018; Li et al., 2024). This process is regulated by nuclear receptor coactivator 4 (NCOA4), a critical mediator of intracellular iron homeostasis. NCOA4 recognizes and binds to ferritin, transporting it to lysosomes for degradation (Latunde-Dada, 2017). This releases stored iron, increasing the pool of bioavailable iron (Mancias et al., 2015). Upregulation of NCOA4 significantly elevates intracellular iron levels, further promoting lipid peroxidation and ferroptosis (Masaldan et al., 2018). Additionally, research has found a close interplay between NCOA4 and other ferroptosis-related signaling pathways. For instance, NCOA4-mediated ferritinophagy may impact the activity of glutathione peroxidase 4 (GPX4), as increased iron levels can deplete intracellular antioxidants like glutathione, indirectly inhibiting GPX4 function and thereby facilitating ferroptosis (Mancias et al., 2014).
Additionally, iron overload in ferroptosis is further modulated by key metal ion transporters, including solute carrier family 39 member 14 (Slc39a14) and solute carrier family 39 member 8 (Slc39a8) (Aierken et al., 2024). Slc39a14 primarily facilitates the uptake of non-transferrin-bound iron (NTBI), significantly contributing to intracellular iron accumulation (Aierken et al., 2024). Likewise, Slc39a8 also has a similar function, enhancing iron import and increasing intracellular iron levels (Aierken et al., 2024). Upregulation of these transporters promotes iron overload, lipid peroxidation, and ferroptosis. Conversely, inhibiting Slc39a14 or Slc39a8 has alleviated ferroptosis by reducing intracellular iron levels and mitigating oxidative stress (Aierken et al., 2024).
Together, the processes of ferritinophagy, mediated by NCOA4, and enhanced iron import via Slc39a14 and Slc39a8 synergistically increase intracellular iron levels, solidifying their roles as critical factors in regulating ferroptosis.
2.1.2 Lipid peroxidationLipid peroxidation, a central driving process of ferroptosis, is a key distinguishing feature from other forms of cell death (Liang et al., 2022). It occurs when reactive oxygen species (ROS) attack polyunsaturated fatty acids (PUFAs) in cellular membranes. ROS, highly reactive molecules derived from molecular oxygen, include superoxide anions, hydrogen peroxide, and hydroxyl radicals. They are primarily generated through enzymatic reactions involving NADPH oxidases (NOXs), cytochrome P450 reductase (POR), and NADH-cytochrome b5 reductase (CYB5R1), with the mitochondrial electron transport chain (mETC) serving as another significant source (Ai et al., 2021; Yan et al., 2021). PUFAs, with their multiple double bonds, are particularly vulnerable to oxidative attack, leading to the formation of lipid peroxidation products (L-OOH) such as malondialdehyde (MDA) and 4-hydroxy-trans-2-nonenal (4-HNE) (Barrera et al., 2015). These toxic byproducts compromise membrane integrity, increase permeability, cause leakage of intracellular components, and trigger inflammatory responses, exacerbating cellular damage (Yang and Stockwell, 2016). Additionally, iron plays a critical role as a cofactor in the biosynthesis of lipoxygenases (LOXs) and cytochrome P450 enzymes, which are central to the generation of ROS during ferroptosis (Zou et al., 2020). Iron metabolism promotes lipid peroxidation through the Fenton reaction (Fe2⁺ + H₂O₂ → Fe³⁺ + ⋅OH + OH⁻), which produces hydroxyl radicals that amplify oxidative stress and accelerate lipid peroxidation, as well as the Haber–Weiss reaction (Dixon et al., 2012).
ROS-driven lipid peroxidation proceeds through three main stages: initiation, propagation, and termination. ROS, such as hydroxyl radicals, abstract allylic hydrogen atoms from PUFAs during initiation, generating lipid radicals (L⋅). In tell as the Haber–Weiss reaction (Dixon et al., 2012), adjacent lipids produce more lipid radicals and lipid hydroperoxides (L-OOH), thus sustaining the chain reaction (Hermetter et al., 2012; Volinsky and Kinnunen, 2013; Que et al., 2018). This process leads to severe oxidative damage if left unchecked, compromising membrane integrity and ultimately triggering ferroptosis.
Notably, the specific composition of membrane phospholipids, particularly the levels of phosphatidylethanolamine (PE) containing polyunsaturated fatty acids (PE-PUFAs), plays a crucial role in determining ferroptosis sensitivity (Kagan et al., 2017). Higher PE-PUFA content increases the susceptibility of cells to lipid peroxidation, thereby elevating the risk of ferroptosis (Kagan et al., 2017). Acyl-CoA synthetase long-chain family member 4 (ACSL4) catalyzes the conversion of PUFAs into PUFA-CoA derivatives, which are then incorporated into PE by lysophosphatidylcholine acyltransferase 3 (LPCAT3) (Kagan et al., 2017). This process enhances membrane susceptibility to oxidative damage, accelerating lipid peroxidation and ferroptosis.
However, lipid peroxidation is not entirely uncontrollable, as multiple regulatory mechanisms exist within cells. Glutathione peroxidase 4 (GPX4), a key inhibitor of ferroptosis, uses glutathione (GSH) as a cofactor to reduce phospholipid hydroperoxides to their corresponding alcohols, thereby protecting cells and membranes from oxidative damage. In addition to the pivotal role of GPX4, recent studies have identified several GPX4-independent pathways that regulate lipid peroxidation and ferroptosis, providing multilayered protection against oxidative stress. Membrane-bound O-acyltransferase 1/2 (MBOAT1/2) is a ferroptosis suppressor (Liang et al., 2023). As a member of the lysophospholipid acyltransferase (LPLAT) family, MBOAT1/2 transfers monounsaturated fatty acids (MUFA) from MUFA-CoA to lysophosphatidylethanolamine (lyso-PE), thereby competitively reducing the incorporation of PUFA-CoA into lyso-PE (Liang et al., 2023). This modification alters the cellular phospholipid profile by increasing PE-MUFA levels while decreasing PE-PUFA levels, the latter being the preferred substrate for lipid peroxidation and a key determinant of ferroptosis sensitivity (Liang et al., 2023). Thus, this selective remodeling decreases the pool of peroxidation-prone phospholipids, offering an alternative pathway to suppress ferroptosis independently of GPX4 and FSP1 (Liang et al., 2023). Moreover, iPLA2β, a member of the calcium-independent phospholipase A2 family, functions independently of glutathione by cleaving oxidized fatty acid chains from phospholipids, directly removing peroxidized products, effectively detoxifying damaged lipids, and maintaining membrane integrity (Chen et al., 2021). This activity is particularly crucial under conditions where GPX4 is absent or impaired, significantly suppressing lipid peroxidation and ferroptosis. Studies have also shown that iPLA2β reduces oxidized phospholipid levels mediated by ALOX12, thereby limiting the accumulation of lipid peroxidation products (Chen et al., 2021). This detoxification mechanism works synergistically with the lipid remodeling functions of MBOAT1 and MBOAT2, forming a multi-layered defense system against ferroptosis.
In summary, lipid peroxidation is central in ferroptosis, driven by ROS-mediated oxidative damage to PUFAs in cellular membranes. While GPX4 is a key regulator in detoxifying lipid peroxides, several GPX4-independent mechanisms, such as lipid remodeling by MBOAT1 and MBOAT2 and lipid detoxification by iPLA2β, provide additional layers of protection. These interconnected mechanisms further illuminate the complexity of ferroptosis regulation and underscore the essential role of lipid peroxidation in its execution.
2.1.3 Collapse of antioxidant defense mechanismsThe antioxidant defense System in cells is critically dependent on glutathione peroxidase 4 (GPX4), a crucial metabolic enzyme that uses glutathione (GSH) to convert lipid peroxides into non-toxic alcohols, thereby minimizing harmful lipid peroxidation and safeguarding cells from ferroptosis. The activity of GPX4 is highly reliant on sufficient intracellular GSH levels, which are synthesized via the cystine/glutamate antiporter System Xc−. Comprising the subunits solute carrier family seven member 11 (SLC7A11) and solute carrier family three member 2 (SLC3A2), System Xc− is a vital antioxidant System, facilitating the 1:1 exchange of cystine and glutamate across the cell membrane (Dixon et al., 2014). The imported cystine is reduced to cysteine within the cell, contributing to GSH synthesis (Dixon et al., 2014). GPX4 converts GSH to oxidized glutathione while reducing toxic lipid peroxides (L-OOH) to their corresponding alcohols (L-OH), thus lowering ROS production (Yang and Stockwell, 2016; Zheng and Conrad, 2020). Under normal physiological conditions, reduced GSH is present in concentrations 10 to 100 times greater than oxidized GSH (GSSG), making the GSH/GSSG ratio a common measure of cellular oxidative stress (Em et al., 2013; Lei et al., 2015; Hambright et al., 2017; Tang et al., 2021). Therefore, maintaining adequate GSH levels and GPX4 activity is crucial for cellular antioxidant defense; any disruption to this mechanism can lead to increased oxidative stress and subsequent cellular damage.
The function of GPX4 is precisely regulated by nuclear factor E2-related factor 2 (NRF2), a key transcription factor that activates the expression of various antioxidant genes, including those encoding GSH synthetase and superoxide dismutase (SOD), facilitating the clearance of excess ROS (Xie et al., 2016; Tang et al., 2021). Under normal conditions, NRF2 is tightly suppressed by Kelch-like ECH-associated protein 1 (KEAP1), which mediates NRF2 degradation through ubiquitination (Kobayashi et al., 2004). However, under oxidative stress, KEAP1 undergoes a conformational change that relieves its suppression, allowing NRF2 to stabilize and upregulate antioxidant gene expression (Sun et al., 2016).
In addition to GPX4 and NRF2, cells employ various antioxidant mechanisms, including SOD, catalase (CAT), and vitamins C and E, to combat oxidative stress (Chen et al., 2016). SOD effectively catalyzes the conversion of superoxide radicals into water and oxygen, serving as the first line of antioxidant defense, while CAT further decomposes hydrogen peroxide to prevent harmful hydroxyl radical formation (Yuewei et al., 2014; Baker et al., 2023). Vitamins C and E enhance cellular antioxidant capacity through their hydrophilic and lipophilic properties, respectively (Tm et al., 2019; Hu Q et al., 2021). However, inflammatory factors such as interleukin-1β (IL-1β) and tumor necrosis factor-alpha (TNF-α) can significantly inhibit SOD and CAT activity, reduce levels of vitamins C and E, and activate the nuclear factor-kappa B (NF-κB) pathway, exacerbating oxidative stress and stimulating inflammatory responses, thereby creating a vicious cycle (Ueda and Takasawa, 2018).
Additionally, the NADPH/FSP1/CoQ10 pathway, through the action of FSP1, reduces CoQ10 with NADPH, further inhibiting lipid peroxidation and ferroptosis (Frei et al., 1990). Research also indicates that the gtp cyclohydrolase 1/Tetrahydrobiopterin/dihydrofolate reductase (GCH1/BH4/DHFR) pathway, particularly DHFR, suppresses ferroptosis by promoting BH4 synthesis; inhibiting DHFR leads to decreased BH4 production, thereby enhancing lipid peroxidation (Soula et al., 2020; Fanet et al., 2021).
Mitochondria, as central organelles for iron, calcium, lipid, and amino acid metabolism, have limited iron content. However, elevated iron levels can disrupt mitochondrial iron homeostasis (Beckman and Ames, 1998). Research has demonstrated that inhibiting mitochondrial tricarboxylic acid cycles or electron transport chains can lower mitochondrial membrane hyperpolarization and lipid peroxidation, underscoring the critical role of mitochondrial pathways in ferroptosis (Steiner and Lang, 2017; Haileselassie et al., 2019).
In summary, the ferroptosis-related biological components discussed above are presented in Table 1 and Figure 1. Figure 1 was created using Adobe Illustrator 2019 (Adobe Inc., San Jose, CA, USA). The design of the signaling pathways related to iron metabolism, the Fenton reaction, lipid peroxidation, and antioxidant pathways was inspired by the overall concepts and graphical elements from the literature to ensure scientific accuracy and rigor (An et al., 2023).

Table 1. Ferroptosis-related biological components in osteoarthritis.
3 The role of ferroptosis in osteoarthritisOsteoarthritis (OA) is characterized by pathological changes affecting the entire joint structure, with degenerative alterations in articular cartilage being the primary manifestation (Findlay and Atkins, 2014). These pathological changes are accompanied by bone spur formation, subchondral bone sclerosis, and synovitis. Under abnormal biomechanical and biochemical stimuli, the balance between anabolic and catabolic processes in chondrocytes is disrupted, leading to the characteristic phenotypic changes associated with OA and promoting disease progression. For example, hypertrophic chondrocytes release matrix degradation products and pro-inflammatory mediators, which aggravate cartilage damage and trigger the proliferation of nearby synovial cells and inflammatory responses (Hunter and Bierma-Zeinstra, 2019; Singh et al., 2019). In turn, these proliferating synovial cells release pro-inflammatory mediators that stimulate the expression of matrix-degrading enzymes and other catabolic factors (Hunter and Bierma-Zeinstra, 2019; Singh et al., 2019). The formation of bone spurs at the joint margins is closely linked to cartilage formation and ossification, a process influenced by inflammatory factors and joint overload (Hsia et al., 2018). Additionally, changes in subchondral bone contribute to cartilage destruction, primarily through an imbalance between osteoblastic and osteoclastic activity (Hu Y et al., 2021).
Recent studies have identified iron overload as a risk factor for osteoarthritis. While research on the relationship between ferroptosis and OA is limited, emerging evidence suggests that ferroptosis plays a crucial role in regulating chondrocyte activity, extracellular matrix degradation, and synovial inflammation, thereby influencing OA progression (Feng et al., 2020; Yao et al., 2021). Elevated iron levels in OA patients’ joint tissues may trigger chondrocyte ferroptosis by enhancing oxidative stress and lipid peroxidation (Feng et al., 2020). In elderly female OA patients, higher iron storage levels may be associated with increased iron burden due to estrogen deficiency. Patients with significantly elevated ferritin levels are more prone to severe joint complications, and this increase correlates positively with the severity of arthritis (Morales-Ivorra et al., 2018; Jing et al., 2021a). However, some studies propose a biphasic effect of iron status on OA progression, where iron overload and deficiency increase OA risk (Kennish et al., 2014). This article will explore how ferroptosis impacts various pathological processes in OA, aiming to provide new insights into its prevention and treatment (Figure 2). Figure 2 was created using Adobe Illustrator 2019 (Adobe Inc., San Jose, CA, USA).
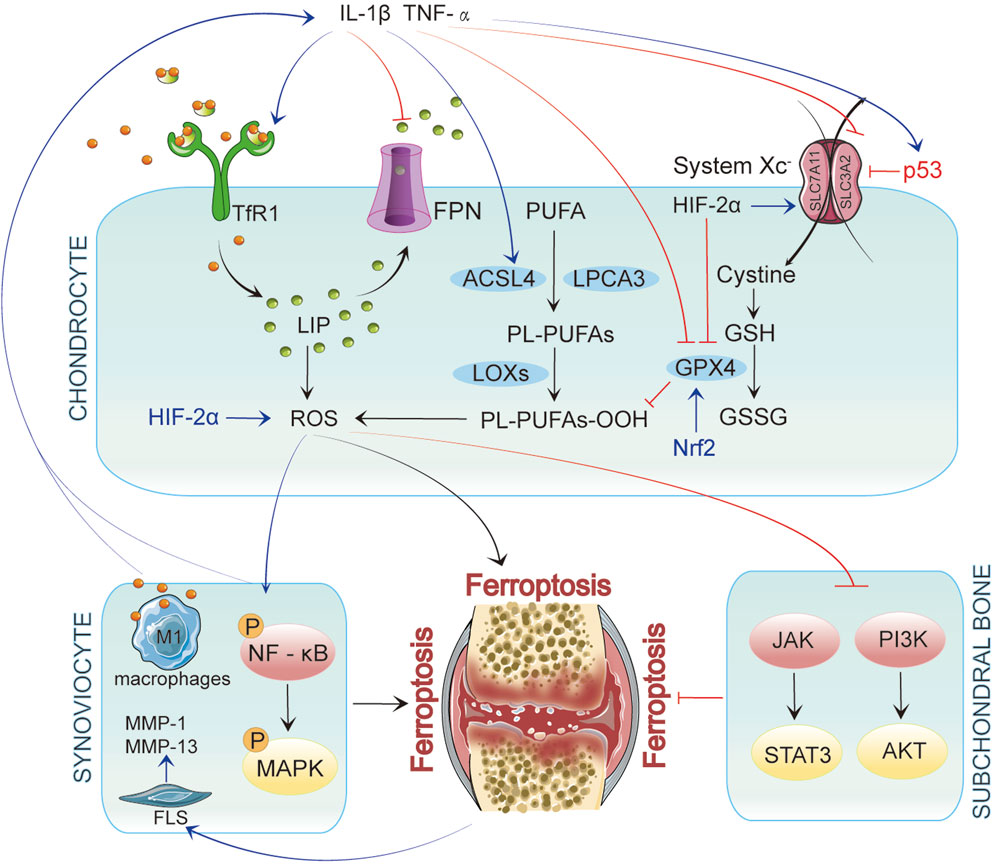
Figure 2. In Chondrocytes, ferroptosis is initiated by iron accumulation and lipid peroxidation. When p53 is upregulated, it inhibits System Xc−, leading to decreased glutathione levels and reduced GPX4 activity, ultimately resulting in ferroptosis. Inflammatory factors such as IL-1β and TNF-α enhance the generation of reactive oxygen species (ROS) by activating TfR1 and ACSL4 while upregulating p53 expression, further promoting ferroptosis. These inflammatory mediators affect chondrocytes and interact with synovial cells and their microenvironment, exacerbating inflammation in OA. In synovial cells, inflammatory signaling pathways activate NF-κB and MAPK, promoting ferroptosis. Concurrently, iron overload polarizes synovial macrophages to the classical M1 type, leading to increased secretion of inflammatory mediators. This activation of inflammation further drives ferroptosis, which activates fibroblast-like synoviocytes (FLSs) to produce matrix metalloproteinases (MMP-1 and MMP-13), intensifying synovitis and creating a vicious cycle. In subchondral bone, ferroptosis is similarly regulated by multiple signaling pathways, particularly the JAK/STAT3 and PI3K/AKT axes.
3.1 Cartilage degeneration and ferroptosisA prominent feature of OA is the degeneration and degradation of articular cartilage, closely linked to the metabolic balance of chondrocytes (Zheng et al., 2021). Chondrocytes, the only cellular component of cartilage, maintain the dynamic equilibrium of the extracellular matrix (ECM) through precise regulation of collagen type II and matrix-degrading enzyme secretion (Chang et al., 2022). However, this balance is disrupted during the pathological progression of OA, leading to metabolic imbalances in chondrocytes and consequent cartilage degradation. Ferroptosis is crucial in the loss of chondrocytes associated with this process.
In mouse models of OA, typical characteristics of ferroptosis have been observed in chondrocytes, including abnormal iron metabolism, lipid peroxidation, and mitochondrial dysfunction (Sun et al., 2021). These alterations impair normal chondrocyte function and exacerbate apoptosis and ECM degradation (Sun et al., 2021). Under iron overload conditions, catabolic pathways in chondrocytes are activated, while anabolic pathways are suppressed, destroying the cartilage matrix and progression of OA. In OA models induced by interleukin-1 beta (IL-1β) and ferric ammonium citrate (FAC), hallmark ferroptosis markers such as glutathione peroxidase 4 (GPX4) and solute carrier family seven member 11 (SLC7A11) exhibit significant downregulation, whereas expression levels of the tumor suppressor gene p53 and acyl-CoA synthetase long-chain family member 4 (ACSL4) are elevated (Yao et al., 2021). These findings further corroborate the involvement of ferroptosis in chondrocytes, indicating mechanisms related to lipid peroxidation and oxidative stress imbalance.
Recent studies have highlighted the mediating role of hypoxia-inducible factor 2 alpha (HIF-2α) in chondrocyte ferroptosis, promoting lipid storage and accumulation of lipid peroxidation products. Overexpression of HIF-2α decreases GPX4 levels and facilitates the activity of carnitine palmitoyltransferase 1A (CPT1A), which transports fatty acids into the mitochondrial matrix for β-oxidation, thus aggravating cartilage degeneration (Zhou et al., 2021).
Moreover, inflammatory cytokines have been found to disrupt iron homeostasis in chondrocytes by upregulating transferrin receptor 1 (TfR1) and downregulating the iron transporter ferroportin (FPN), contributing to iron overload (Jing et al., 2020). In OA, This iron overload is closely related to disease progression, as it not only directly induces chondrocyte apoptosis but also accelerates cartilage degradation by upregulating matrix-degrading enzyme expression (Jing et al., 2021a). For instance, iron overload induced by ferric ammonium citrate significantly enhances chondrocyte apoptosis and increases the expression of matrix metalloproteinases (MMP3, MMP13) (Jing et al., 2021a). Furthermore, downregulation of nuclear factor E2-related factor 2 (Nrf2) leads to reduced GPX4 expression, thereby intensifying ferroptosis (Ni et al., 2020; Lu et al., 2022; Yang et al., 2022b). As a key antioxidant transcription factor, Nrf2’s interaction with ferroptosis illustrates its critical role in regulating oxidative stress and cellular fate.
Beyond directly disrupting iron homeostasis, ferroptosis further contributes to the pathological progression of OA through additional mechanisms. The roles of HIF-2α, GPX4, and lipid peroxidation in ferroptosis are particularly critical. Ferroptosis damages chondrocytes directly and exacerbates joint injury by inducing local inflammatory responses. For instance, iron overload can enhance chondrocyte catabolism through overactivated NLRP3 inflammasomes, promoting MMP expression and exacerbating cartilage degeneration (Li S et al., 2023).
Recent studies have established a close relationship between ferroptosis and ECM degradation (Wilkinson et al., 2019). The ECM, composed of proteoglycans, collagen, and minerals, is essential for maintaining the biomechanical properties of articular cartilage. However, as chondrocyte function declines, tissue hydration decreases, leading to progressive ECM damage. In the early stages of OA, proliferating chondrocytes disrupt ECM composition, resulting in reduced proteoglycan synthesis and altered matrix permeability and mechanical compliance (Sun et al., 2012). Inflammatory factors and proteases play pivotal roles in ECM degradation. Evidence suggests that iron regulates ECM deposition and remodeling via oxidative stress, indicating its critical role in ECM integrity (Maldonado and Nam, 2013; Wilkinson et al., 2019). Additionally, mechanical injuries, such as joint wear, trigger inflammatory responses in cartilage and synovium, further upregulating the expression of matrix-degrading proteins, accelerating ECM degradation, and promoting OA progression (Maldonado and Nam, 2013). Studies have shown that iron can enhance IL-1β-induced expression of MMP-3 and MMP-13, further facilitating ECM degradation (Maldonado and Nam, 2013). This association with iron accumulation in chondrocytes is characterized by IL-1β upregulating TfR1 and DMT1 while downregulating FPN1, which exacerbates iron overload, disrupts ECM balance, and ultimately leads to cartilage damage (Jing et al., 2020; Jing et al., 2021). While the negative impact of iron overload on ECM has been established, the specific mechanisms by which ferroptosis contributes to ECM degradation remain underexplored. Given that ECM degradation involves inflammation and oxidative stress—key components of ferroptosis—a complex relationship between ferroptosis and ECM degradation warrants further investigation.
In recent years, the roles of oxidative stress and mitochondrial dysfunction in the progression of OA have garnered significant attention (Blanco et al., 2011; Suantawee et al., 2013; Ansari et al., 2020; Wang et al., 2020; Zheng et al., 2021). Excessive iron can induce mitochondrial dysfunction in chondrocytes by producing reactive oxygen species (ROS), thereby promoting the expression of OA catabolic markers (Jing et al., 2021a). However, the implications of iron-induced mitochondrial dysfunction on chondrocyte metabolism require deeper exploration (Jing et al., 2021b). Additionally, studies suggest that the mitochondrial membrane potential in chondrocytes decreases significantly impacts mitochondrial structure and function (Jing et al., 2021b). Under IL-1β stimulation, chondrocyte mitochondrial membrane potential decreases substantially, closely correlating with mitochondrial dysfunction (Chen B. Y et al., 2024). The interconnections among ferroptosis, oxidative stress, and mitochondrial dysfunction in OA cartilage degeneration collectively influence chondrocyte metabolism, ECM balance, and the progression of OA. Nonetheless, these three elements’ specific regulatory networks and mechanisms require further elucidation.
3.2 Synovial inflammation and ferroptosisSynovial inflammation is another critical feature in the pathological progression of OA and may even precede cartilage degeneration (Scanzello and Goldring, 2012). Synovial changes are closely linked to symptoms such as pain and functional impairment. Research suggests that iron homeostasis in synovial tissue is regulated by iron regulatory proteins, revealing a strong link between synovitis, synovial hyperplasia, and iron deposition (Nieuwenhuizen et al., 2013). For instance, in patients with hemophilia, significant deposits of hemosiderin in the periosteum can stimulate fibroblast proliferation, leading to hemophilic synovitis (Wen et al., 2002; Nieuwenhuizen et al., 2013). Synovial lesions mainly impact areas near cartilage degeneration and are marked by synovial lining cell hyperplasia, infiltration of inflammatory cells, neovascularization, and fibrin deposition (Sellam and Berenbaum, 2010).
Recent research progressively highlights the potential role of ferroptosis in synovial cells as a major factor contributing to synovial inflammation. Compared to healthy individuals, OA patients exhibit significantly elevated levels of iron ions in synovial fluid, alongside observable iron deposits in synovial tissue (Yazar et al., 2005). Under conditions of iron overload, macrophages in the synovium polarize into classically activated M1 macrophages (Hakobyan et al., 2004; Nieuwenhuizen et al., 2013; Zhou et al., 2018; Wu et al., 2020). This process is closely associated with ROS production induced by iron overload and p53 acetylation (Stockwell et al., 2017). M1 macrophages secrete pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, which trigger further inflammatory responses in the synovium and contribute to cartilage destruction and osteophyte formation, thus exacerbating OA progression (Marchev et al., 2017; Ansari et al., 2020). Ferroptosis induced by iron overload may also disrupt the synovial microenvironment, impairing the function of fibroblast-like synoviocytes (FLS). Ferroptosis can trigger a non-specific immune response, leading to the release of IL-1β, which activates FLS to produce MMP-1 and MMP-13—proteins closely associated with synovial inflammatory responses (Ni et al., 2018; Jing et al., 2021a). Additionally, IL-1β promotes the expression of cyclooxygenase-2 (COX-2) in FLS, enhancing the secretion of prostaglandin E2 (PGE2) and further aggravating synovial inflammation (Tsai et al., 2017).
The release of lipid peroxidation products and oxidative stress signals during ferroptosis activates inflammatory pathways in synovial cells, such as the NF-κB and MAPK pathways, resulting in the release of additional inflammatory factors and creating a vicious cycle (Kumar et al., 2019; Tm et al., 2019; Liu et al., 2022; Lv et al., 2022; Miao et al., 2022). ACSL4 is highly expressed in OA synovial tissue, while the key antioxidant enzyme GPX4 expression is significantly reduced. ACSL4 influences the oxidative-antioxidative balance within cells, promoting ferroptosis, whereas the deficiency of GPX4 exacerbates ferroptosis and associated inflammatory responses (He W et al., 2023; Zheng et al., 2024).
Moreover, an imbalance in the exchange of glutamate and cystine in the System Xc− transport system inhibits its function, leading to glutathione depletion and indirect suppression of GPX4 activity, thus facilitating ferroptosis. Glutamate may also induce intra-articular inflammatory responses and abnormal pain in OA patients by activating N-methyl-D-aspartic acid (NMDA) receptors, further advancing OA progression (Piepoli et al., 2009).
3.3 Subchondral bone remodeling and ferroptosisSubchondral bone remodeling is a hallmark pathological feature in advanced stages of OA, typically characterized by bone sclerosis and osteophyte formation. Research indicates a close interplay between articular cartilage and subchondral bone. On the one hand, abnormal remodeling and angiogenesis in subchondral bone can destroy articular cartilage. On the other hand, chondrocytes can regulate subchondral bone remodeling through the RANK-RANKL-OPG signaling pathway (Hu Y et al., 2021). Mechanistically, the core of bone remodeling lies in the balance between osteoblasts’ bone-forming activity and osteoclasts’ bone-resorbing activity (Simao and Cancela, 2021). The death of chondrocytes and the cartilage matrix’s degradation can disrupt the cartilage-bone interface’s structure, consequently affecting osteoblasts’ and osteoclast activity (Miao et al., 2022). In this process, ferroptosis plays several critical roles (Miao et al., 2022).
Studies have shown that iron overload elevates intracellular ROS levels, triggering lipid peroxidation, disrupting redox balance, and altering the bone marrow microenvironment (Cen et al., 2018). This process blocks the JAK/STAT3 and PI3K/AKT signaling pathways, activates the p38-MAPK pathway, and induces G1 cell cycle arrest and autophagy in osteoblasts (Cen et al., 2018). Elevated ROS also promotes osteoclast differentiation via NF-κB, JNK, and ERK signaling, driving abnormal bone remodeling and OA progression (Wang et al., 2018). Furthermore, iron overload can directly impair osteoblasts’ and osteoclast functions and alter the bone matrix’s composition and structure, facilitating bone sclerosis and osteophyte formation. This abnormal bone remodeling further increases the mechanical burden on the joint, exacerbating OA symptoms (Hu Y et al., 2021).
Additionally, hypoxia-inducible factor 1 (HIF-1) is a key regulator of chondrocyte growth and survival, significantly influencing the state of subchondral bone. Research indicates that HIF-1α-induced long non-coding RNA (lncRNA) PMAN can regulate ferroptosis by maintaining the mRNA stability of the ferroptosis-related gene SLC7A11, inhibiting ferroptosis and contributing to subchondral bone remodeling (Fan et al., 2018; Lin et al., 2022). This interaction affects the progression of osteoarthritis. However, the specific roles of ferroptosis-related genes and miRNAs remain to be explored further, representing a potential direction for future research into ferroptosis.
In conclusion, the biological molecules of ferroptosis involved in this part are shown in Tables 1, 2.
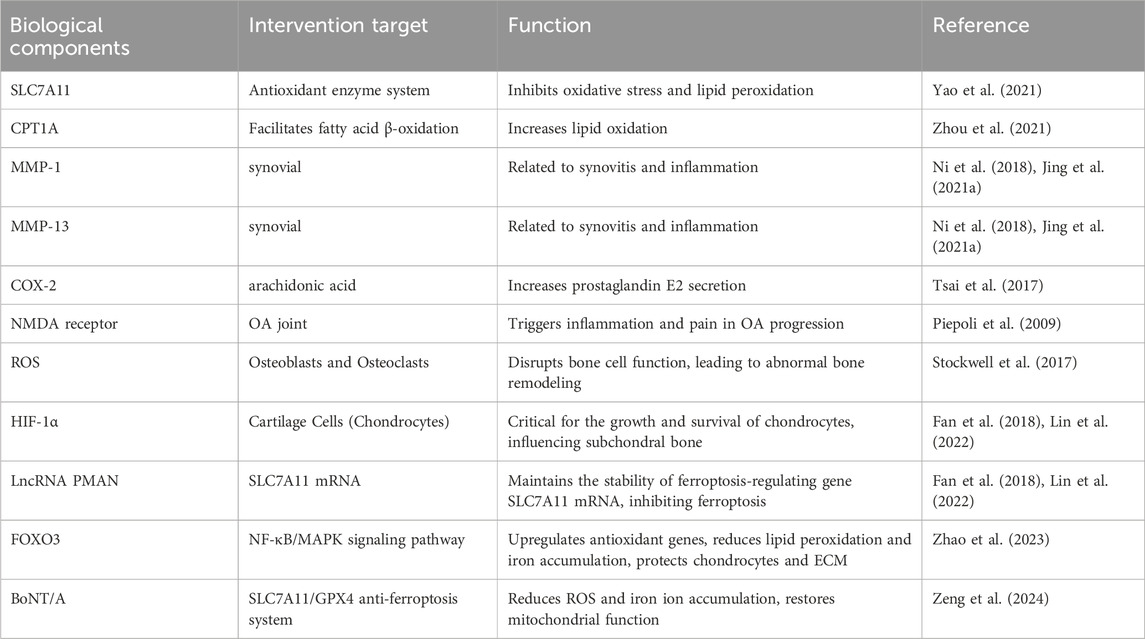
Table 2. Ferroptosis-related biological components in osteoarthritis.
4 Biomarkers related to ferroptosis and clinical prospects in osteoarthritisProgressive joint pain is a hallmark of osteoarthritis (OA), and treatments aim to alleviate pain and improve joint function. Conventional treatments encompass lifestyle changes, physical therapy, medication, and surgical interventions, with effectiveness depending on disease severity and duration, highlighting the need for individualized treatment. New therapies such as nerve blocks, mesenchymal stem cell injections, platelet-rich plasma injections, and osteoporosis medications like strontium ranelate offer additional options for OA management. However, due to the incomplete understanding of OA’s pathogenesis, recognized effective treatment methods remain limited.
Iron homeostasis imbalance, leading to iron overload and subsequent ferroptosis, can damage the synovium, cartilage, and extracellular matrix (ECM), playing a crucial role in OA progression. Key proteins and genes involved in iron metabolism, lipid peroxidation, and associated signaling pathways may be potential therapeutic targets for OA. Ferroptosis-related molecular biomarkers hold great potential for diagnosing OA, monitoring disease progression, and guiding personalized treatment strategies. The following sections will delve into biomarkers associated with ferroptosis, iron metabolism, lipid peroxidation, and antioxidant defenses, analyzing their clinical application prospects in OA.
4.1 Iron metabolism-related biomarkers and osteoarthritis4.1.1 Transferrin receptor 1 (TfR1)TfR1 expression is elevated in OA patients’ synovial tissue and chondrocytes, making it a potential early diagnostic marker that reflects disease progression. Its integration with imaging techniques can enhance diagnostic accuracy, facilitating timely intervention. Research on TfR1 has identified several therapeutic interventions. For example, ferristatin II promotes the degradation of TfR1, disrupting Tf-mediated iron delivery and intervening in the pathological processes of OA (Yuewei et al., 2014). Additionally, Biochanin A (BCA) can reduce intracellular iron levels by suppressing TfR1 activity and enhancing the expression of ferroportin (FPN), thereby alleviating mitochondrial damage and chondrocyte apoptosis induced by iron overload (He Q et al., 2023).
Iron chelators represent an effective intervention for patients with excessive TfR1 expression, binding circulating and intracellular iron for excretion via urine or bile, thereby reducing ferroptosis (Di Maggio and Maggio, 2017). Commonly used iron chelators include deferoxamine (DFO), deferiprone (DFP), and deferasirox (DFS) (Rodrigues de Morais and Gambero, 2019). In vitro studies have demonstrated that DFO can inhibit the degradation of type II collagen in osteoarthritis cartilage while also reducing the expression of inflammatory factors such as MMP-1, MMP-13, IL-1β, and TNF-α (Tchetina et al., 2016). Furthermore, DFO can restore ferritin levels by inhibiting NCOA4 expression and chelating excess iron to regulate iron homeostasis. These findings indicate that iron chelators regulate iron levels and contribute to managing inflammation associated with OA.
However, existing iron chelators exhibit side effects in clinical applications, including gastrointestinal discomfort, hepatotoxicity, and nephrotoxicity (Guo et al., 2024). Thus, developing novel iron chelators, particularly nanoscale formulations, may represent a future direction to reduce side effects while maintaining or enhancing therapeutic efficacy (Kalanaky et al., 2016). Additionally, the compound White Cardamonin (CAR), derived from Vitex negundo, protects cartilage by mitigating iron overload-induced chondrocyte damage and apoptosis (Li S et al., 2023). CAR activates the SIRT1 pathway and inhibits the p38 MAPK signaling pathway, reducing NLRP3 inflammasome production and alleviating inflammation and cartilage degeneration (Li S et al., 2023). It also reverses the reduction in type II collagen expression and the elevation of MMPs caused by iron overload, thereby mitigating chondrocyte degeneration (Li S et al., 2023).
Based on TfR1-mediated iron uptake and ferroptosis mechanisms, researchers are exploring possibly delaying OA progression by intervening in ferroptosis. Inhibiting TfR1 expression may reduce chondrocyte ferroptosis and diminish synovial inflammation, slowing OA advancement. Currently, ferroptosis inhibitors such as Ferrostatin-1 and Liproxstatin-1 have entered clinical trials, potentially leading to precision treatment strategies that integrate TfR1 detection techniques (Li S et al., 2023).
Despite the promising clinical prospects of TfR1 in OA, challenges remain in its practical application. Future research should further validate the specificity and sensitivity of TfR1 as an early diagnostic marker and develop cost-effective detection technologies. Moreover, multi-omics analyses could help elucidate the interactions between TfR1 and other ferroptosis regulatory molecules, such as GPX4, providing a comprehensive perspective to support personalized treatment strategies.
4.1.2 Divalent metal ion transporter 1 (DMT1)DMT1 plays a crucial role in the transport of iron ions, and its abnormal expression in OA patients is closely associated with disease progression. Research has shown that the upregulation of DMT1 results in excessive iron accumulation within chondrocytes, increasing the risk of cellular damage (Li S et al., 2023). Research indicates that DMT1 is critical in the progression of osteoarthritis (OA) driven by iron overload; inhibiting DMT1 alleviates IL-1β-induced inflammatory responses and extracellular matrix degradation by significantly suppressing the MAPK and PI3K/AKT/NF-κB signaling pathways (Liu et al., 2024). This finding suggests that regulating the activity of the iron transporter DMT1 could offer a novel therapeutic target for OA.
Implementing DMT1 inhibitors as an intervention strategy demonstrates significant therapeutic potential (Jing et al., 2020). Additionally, multi-target combination strategies are gaining attention. For instance, combining DMT1 inhibitors with antioxidants can yield synergistic effects across multiple pathways. This approach reduces iron ion accumulation, suppresses ferroptosis, and enhances antioxidant defense mechanisms, thereby protecting chondrocytes from oxidative stress damage and effectively slowing OA progression.
4.1.3 FerritinFerritin consists of heavy chain ferritin (FTH1) and light chain ferritin (FTL), and it primarily functions to store intracellular iron, preventing excessive accumulation of free iron and reducing cellular iron toxicity (Arosio et al., 2017). In OA patients, alterations in ferritin expression and stability lead to elevated levels of free iron, heightening the risk of ferroptosis. Studies have shown that in models of iron overload and destabilization of the medial meniscus (DMM)-induced OA, FTH1 expression is significantly elevated, accompanied by increased ferritin levels in the synovial tissue (Yuan et al., 2024). Thus, monitoring changes in both FTH1 and FTL may provide new insights for early OA diagnosis.
Combining the detection of ferritin with other ferroptosis biomarkers, such as TfR1, DMT1, and GPX4, holds promise for enhancing diagnostic sensitivity and accuracy, thereby offering stronger support for early intervention. Additionally, variations in ferritin levels may serve as auxiliary indicators for evaluating the efficacy of antioxidant treatments (Arosio et al., 2017). The concurrent use of antioxidants, such as N-acetylcysteine (NAC), may improve treatment precision and reduce adverse effects (de Andrade et al., 2015; Li et al., 2022).
Furthermore, ferritin plays a dual role in iron metabolism imbalance; while it helps sequester excess free iron, it may also act as a source of free iron during ferroptosis (Bauckman et al., 2015; Bellelli et al., 2016). Research has identified YL-939, a ferroptosis inhibitor targeting prohibitin 2 (PHB2), which can reduce iron levels in the labile iron pool (LIP) by modulating ferritin expression and uptake (Yang et al., 2022a). Consequently, in future developments of ferroptosis inhibitors, ferritin is anticipated to be a potential target. Its combination with iron chelators and agents that regulate ferritin could further enhance intervention strategies for ferroptosis, providing new avenues for OA treatment.
4.1.4 Ferroportin (FPN)FPN is mammalian cells’ sole iron exporter, pivotal in maintaining intracellular iron homeostasis. Recent studies have highlighted the significance of FPN in regulating ferroptosis. The degradation of FPN, mediated by the E3 ubiquitin ligase RNF217, leads to intracellular iron accumulation, thereby increasing susceptibility to ferroptosis (Jiang et al., 2021). Therefore, maintaining proper FPN function is crucial for preventing iron overload and subsequent ferroptotic cell death in joint tissues.
Therapeutic approaches to prevent FPN degradation may hold promise for mitigating OA progression. For instance, targeting the RNF217-mediated degradation pathway of FPN could help preserve its function, thereby reducing intracellular iron accumulation and the risk of ferroptosis. Additionally, combining such approaches with antioxidants or ferroptosis inhibitors might synergize in protecting chondrocytes and synovial cells from iron-induced damage (Jiang et al., 2021).
Further research is needed to elucidate the precise role of FPN in OA and to develop targeted therapies that modulate its expression or activity. Understanding the interplay between iron metabolism, FPN regulation, and ferroptosis will be essential in devising effective interventions for OA and other iron-related pathologies.
4.1.5 Hepcidin and NCOA4Hepcidin and NCOA4 play crucial roles in regulating iron homeostasis and ferroptosis, and their abnormal expression is closely linked to the progression of OA (Bellelli et al., 2016; Wang and Babitt, 2019). Hepcidin, a central regulatory hormone in iron metabolism, promotes the degradation of FPN, leading to decreased extracellular iron export and subsequent intracellular iron overload (Wang and Babitt, 2019). This mechanism exacerbates chondrocyte damage associated with ferroptosis in OA patients. Concurrently, NCOA4 mediates the autophagy of ferritin, releasing stored iron and further increasing free iron levels within cells (Bellelli et al., 2016). This process induces oxidative stress and lipid peroxidation, accelerating ferroptosis and driving the progression of OA.
Targeting these mechanisms by inhibiting the activity of hepcidin and NCOA4 offers new therapeutic avenues for OA. Recently, a class of ferroptosis inhibitors that target NCOA4 has been identified. These compounds bind to NCOA4, disrupting its interaction w
留言 (0)