
記住我

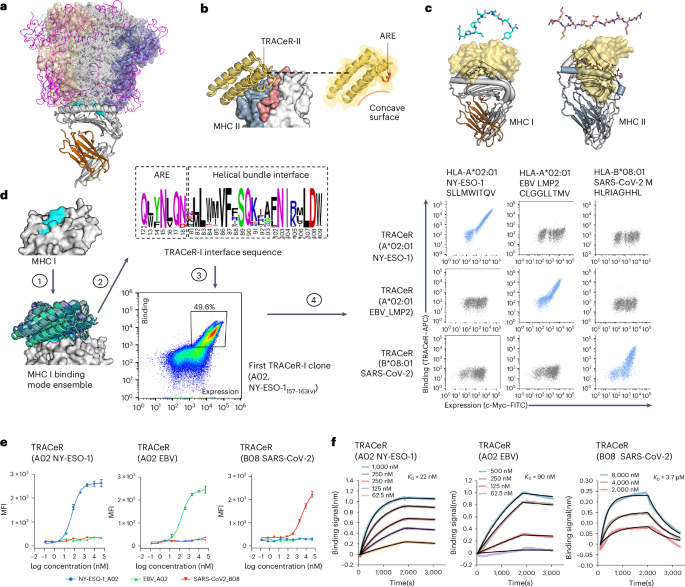


We modeled TRACeR-I in four steps. In the initial step, we use global docking algorithms to model the TRACeR–MHC I interaction in silico at low-resolution, aiming to identify potential docking poses that align with the TRACeR backbone structure. Starting from the available crystal structure to guide the modeling and design (PDB 1S9W (ref. 36)), we used a low-resolution docking algorithm, Patchdock37, to seed the search trajectories of a high-resolution algorithm, RifDock35. From the RifDock output, a cluster of 30 compatible docking modes with large footprint on the peptide antigen between TRACeR and MHC I were identified. In contrast to TRACeR-II’s binding mode, in which the helical bundle binds perpendicular to the antigen groove, RifDock models favored alternative orientations in which the helical bundle lies in parallel to the groove. We then carried out calculations to design the sequences to these models using an iterative protocol established in the Rosetta modeling suite38,39,40. An iterative Rosetta high-resolution docking and interface sequence design protocol was applied on top of the RifDock outputs39,40, with the intention of identifying diverse amino acid substitutions on the interface that are compatible with a given docking pose. Outputs that passed all filtering metrics were ranked on the basis of binding energy (ΔΔG). The top 1,000 outputs were selected and implemented for experimental test in a combinatorial library. The selected interface positions and their corresponding sequences were integrated to create a combinatorial library using Swiftlib41, which optimizes degenerate codon coverage by taking into account a given set of allowed amino acids at each position. Docking and design scripts and evaluation metrics are included in Supplementary Appendices 1–6.
Library design and productionCombinatorial DNA libraries (libraries 1 and 2) were constructed from assembly PCR using Ultramer oligonucleotides (Integrated DNA Technologies) to encode the variable region. Library sequences are listed in Supplementary Table 1.
For library transformation, Saccharomyces cerevisiae yeast EBY100 cells were transformed with insert DNA and linearized pCTCON2 plasmid using an established protocol64. After transformation, cells were grown overnight in SDCAA medium at 30 °C, passaged once and stored in 20% glycerol solution at −80 °C.
Yeast display and library screeningTransformed yeast cells were grown in SDCAA medium. For induction of expression, yeast cells were centrifuged at 2,000g for 5 min and resuspended in SGCAA medium supplemented with 0.2% glucose at a cell density of 1 × 107 cells per ml and induced at 30 °C for 16–24 h. Cells were washed with PBSA (PBS with 0.5% BSA) and labeled with pMHC I tetramer or monomers, together with anti-c-Myc fluorescein isothiocyanate (FITC; Miltenyi Biotech). After incubation for ~1 h at room temperature (RT), cells were washed twice, resuspended in PBSA and then run on a Sony SH800 cell sorter. NGS of final enriched pools was performed with the Azenta Amplicon-EZ service.
The theoretical complexity for screening out initial binders in this background library (library 1) was 1.3 × 1011, but our experimental screening coverage sampled a subset of the theoretical library with 1 × 109 transformed variants. Nonetheless, after five rounds of FACS against HLA-A*02:01/NY-ESO-1, one single clone was enriched from the initial pool of a binding population. The theoretical complexity for the ARE library (library 2) was 1.28 × 109; for each target, 1 × 108 cells were sorted through FACS. For the master library derived from the monomeric TRACeR design, the theoretical library size was 198 = 1.7 × 1010 (eight randomized positions; each position had 19 amino acids without cysteine). For each target, 2 × 109 cells were processed during the first round of magnetic-activated cell sorting.
Binder protein expression and purificationGenes encoding the designed protein sequence were synthesized and cloned into pET-24a(+) Escherichia coli plasmid expression vectors (Genscript, C-terminal 6xHis-tag). Plasmids were then transformed into chemically competent BL21(DE3) E. coli (Zymo Research). The cells were cultured in 2xYT medium at 37 °C until the optical density (OD) reached 0.6–0.8. Protein expression was then induced with 1 mM IPTG at 16 °C. After overnight expression, cells were collected and resuspended with 50 mM Tris buffer pH 8.0 and 300 mM NaCl and frozen at −80 °C until extraction and purification. The cell pellet was thawed and sonicated and purified by nickel affinity followed by SEC (Superdex 75 10/300GL, GE Healthcare). All protein samples were characterized by SDS–PAGE. Protein concentrations were determined by absorbance at 280 nm measured with a Nanodrop spectrophotometer (Thermo Fisher Scientific) using the predicted extinction coefficient.
CDCD spectra were measured on a JASCO CD spectrophotometer in a 1-mm-pathlength cuvette (Hellma). Protein samples were at ~0.2 mg ml−1 in the 50 mM Tris buffer. Melting temperature ranged from 20 to 95 °C and the absorption signal was monitored at 222 nm in 1 °C increments per minute, with 10 s of equilibration time and 1 s of digital integration time. Wavelength scans (200–260 nm) were collected at 20 and 95 °C and again at 20 °C after fast refolding.
BLIBLI binding data were collected on an Octet QK (ForteBio) and processed using the instrument’s integrated software. For binding assays, biotinylated pMHC Is were loaded onto streptavidin-coated biosensors (ForteBio) at 1.25 μg ml−1 in the kinetic buffer (ForteBio) for 1,800 s. Analyte proteins were diluted from concentrated stocks into the binding buffer. After baseline measurement in the binding buffer alone, the binding kinetics were monitored by dipping the biosensors in wells containing the target protein at the indicated concentration (association step) and then dipping the sensors back into baseline or buffer (dissociation). The association time was 1,800 s and dissociation time was 1,200 s, with a shaking speed of 1,000 rpm. Data were analyzed and processed using ForteBio Data Analysis software 9.1.0.
Peptide librariesAll peptide sequences are given as standard single letter codes. Peptides were purchased from Genscript or Mimotope at a purity of >90%. The placeholder peptide gTAX (LFGYPVYV) was purchased from Genscript at a purity of 98%. Peptides were solubilized in a solution of less than 5% v/v DMSO at a concentration of 100 μM. All peptide solutions were centrifuged at 14,000 rpm for 15 min and filtered before use.
Recombinant MHC protein expression, refolding and purificationPlasmid DNA encoding the BirA substrate peptide (BSP, LHHILDAQKMVWNHR)-tagged luminal domain of the MHC I heavy chains and human β2-microglobulin (β2m) were provided by the National Institutes of Health (NIH) tetramer facility (Emory University) and transformed into E. coli BL21(DE3) cells (Novagen). BSP-tagged MHC I proteins were expressed in Luria–Bertani medium and inclusion bodies were collected and purified using a standard protocol51,52. In vitro refolding of BSP-tagged pMHC I molecules was performed by slowly diluting a 200-mg mixture of BSP-tagged MHC I and β2m at a 1:3 molar ratio over 24 h in refolding buffer (0.4 M l-arginine, 100 mM Tris pH 8, 2 mM EDTA, 4.9 mM reduced glutathione and 0.57 mM oxidized glutathione) containing 10 mg of the placeholder peptide. BSP-tagged pMHC I refolding proceeded for 96 h and was followed by SEC for protein purification.
Biotinylation and tetramer library preparationBiotinylation of various pMHC Is and tetramer library preparation using gTAX/HLA-A*02:01 protein were performed as previously described52. The BSP-tagged pMHC I proteins were biotinylated using the BirA biotin–protein ligase bulk reaction kit (Avidity) according to the manufacturer’s instructions and prepared at a final concentration of 2 mg ml−1 monomer. The level of biotinylation was evaluated by SDS–PAGE gel shift assay in the presence of excess streptavidin.
For tetramer library preparation, biotinylated gTAX/HLA-A*02:01 were mixed with TAPBPR (7:1 molar ratio of gTAX-loaded HLA-A*02:01 to TAPBPR) and individual peptides from TCGA peptide library or NY-ESO positions 1, 4, 5, 7 and 8 full-scanning peptide libraries (1:10 molar ratio of gTAX-loaded HLA-A*02:01 to peptide). Each reaction was incubated 2 h at RT and the peptide exchange reactions were confirmed by differential scanning fluorimetry. Meanwhile, streptavidin–(R)-phycoerythrin (PE) or streptavidin–allophycocyanin (APC) (Agilent Technologies) at a 4:1 molar ratio of monomer to streptavidin was added to HLA-A*02:01 proteins in the presence of excess peptides every 10 min over ten intervals at RT in the dark. Tetramerized molecules upon peptide exchange were washed using Amicon Ultra centrifugal filter units with a 100-kDa membrane cutoff to remove excess peptides and TAPBPR with a 1:1,000 dilution of PBS buffer. Biotinylated pMHC I proteins, which did not require exchange for TCGA and NY-ESO-1 positions 1, 4, 5, 7 and 8 full-scanning peptide libraries, were prepared by directly adding streptavidin–PE or streptavidin–APC at a 4:1 molar ratio of monomer to streptavidin every 10 min over ten intervals at RT in the dark. The resulting tetramers could be stored at 4 °C for up to 4 weeks.
Differential scanning fluorimetry was used to access the thermal stabilities of the peptide-exchanged pMHC I molecules and validate the peptide loading. Individual peptide-exchanged HLA-A*02:01 protein was mixed with 10× Sypro orange dye in a buffer of 150 mM NaCl and 20 mM sodium phosphate pH 7.4 to a final volume of 70 µl. Samples were loaded into MicroAmp Optical 384-well plate. The experiment was performed on a QuantStudio 5 real-time PCR machine with excitation and emission wavelengths set to 470 nm and 569 nm. The temperature was incrementally increased at a rate of 1 °C per minute between 25 and 95 °C. Data analysis and fitting were performed in GraphPad Prism 9.
SSM scan on NY-ESO-1157–165(V) peptide using pMHC I tetramersTRACeR was expressed on the yeast cell surface and double-stained with anti-C-myc FITC (Miltenyi Biotech) and MHC tetramer PE. Furthermore, 1G4 TCR was expressed on the T cell surface and double-stained with anti-TCR antibody AlexaFluor 488 and MHC tetramer PE. We used the chaperone-mediated peptide exchange method to load individual antigen variants into HLA-A*02:01 conjugated to fluorescently labeled tetramers51,52 and detected binding patterns by flow cytometry. After 30 min of incubation, cells were washed with PBSA twice and analyzed on flow cytometry. The median fluorescence intensity (MFI) of TRACeR was calculated on the basis of the FITC-positive population. The MFI of 1G4 TCR was calculated on the basis of the TCR-positive population.
$$}\;}}=\left(\frac}_}}}_}-}-1_}}\right)\times 100$$
Sequence-based selection of cross-reactive peptidesWe searched the Immune Epitope Database65 for linear peptides that were positive for binding to HLA-A*02:01 (72,900 peptides) with the SMW motif using the following regular expression: ‘[SCT].MW’. Three peptides (FTIAMWLLL, CINMWCWTV and TSDMWLYR) had known protein origin and the SMW motif in the middle of the peptide and were selected for further analysis.
Triplet NNK library on yeast displayed pMHC I with NY-ESO-1 peptideNY-ESO-1/HLA-A*02:01 was expressed on the surface of YVH10 yeast using a single-chain format consisting of peptide, B2M and the MHC I alpha chain ecotodomain, separated by GS linkers. A V5 tag was included at the C terminus of the MHC I alpha chain for detection of surface display. A peptide SSM scanning library was generated by diversifying one to three amino acid positions using degenerate NNK codons spanning the length of the NY-ESO-1 peptide. The library was built using homologous recombination of peptide oligo pools (Integrated DNA Technologies) with a vector encoding the B2M and MHC I alpha chain using electroporation conditions described previously66. The scanning library had a theoretical nucleotide diversity of 6.7 × 105.
Transformed yeast were grown in SDCAA uracil dropout medium. Yeast cells were induced for pMHC expression by inoculating freshly passaged cells at an OD at 600 nm of 0.1 per ml in SGCAA medium, ensuring coverage of the nucleotide diversity by at least tenfold. Cells were induced at 30 °C with 200 rpm shaking overnight. To label yeast with TRACeR or 1G4 TCR, cells were washed with staining buffer (1× PBS with 1% BSA) before incubating with biotinylated TRACeR or 1G4 tetramer for 1 h at RT. Tetramers were generated by combining soluble 1G4 TCR expressed recombinantly with a C-terminal SpyTag and recombinant SpyCatcher-Fc fusion at a 4:1 molar ratio for 20 min at RT. Cells were washed twice using staining buffer before labeling with 1 µg ml−1 of AlexaFluor 405-conjugated anti-V5 antibody and either 1:500 dilution of streptavidin–APC-conjugated (Invitrogen) or 1:100 dilution of streptavidin–PE-conjugated anti-human Fc antibody (Biolegend) for TRACeR or 1G4, respectively, in staining buffer for 20 min on ice. Next, cells were washed twice, resuspended in staining buffer and sorted on the FACS Melody (BD Biosciences). Yeast cells expressing HLA-A*02:01/NY-ESO-1 were first stained with biotinylated TRACeR in titrating concentrations ranging from 0.25 to 1,000 nM to interpolate an EC80 binding concentration of 156 nM to use for FACS selections. Triplet scanning yeast libraries were stained with 156 nM TRACeR or 100 nM 1G4 tetramer for all sorts. Deep sequencing of sorted libraries was performed as previously described67. Briefly, plasmids were extracted from yeast and a ~150-bp stretch of DNA spanning the peptide was PCR-amplified. DNA libraries were barcoded with xGen UDI primers (Integrated DNA Technologies) in a second round of PCR and prepared for sequencing on the Element AVITI (Element Biosciences).
Immunogenicity assayMice were maintained in a specific-pathogen-free facility at the University of Pennsylvania. Experiments and procedures were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania (protocol 803042). Female BALB/c mice from Jackson Labs, aged 5–7 weeks (n = 8 per group), were randomly assigned to different groups. They were anesthetized and administered Tris buffer through retro-orbital injection. The injection included 50 mM Tris buffer at pH 8 or mAbs mIgG (Innovative IR-MSBC-GF) or hIgG (Innovative IR-HU-GF-ED) at a dose of 0.2 mg kg−1. Injections were administered every 2–3 days over the period of 4 weeks on days 0, 2, 4, 7, 9, 11, 14, 16, 18, 21, 23, 25 and 28. Blood samples were collected on days 0, 8, 15, 22 and 29 through retro-orbital bleeding using micro-hematocrit capillary tubes (Fisher).
Preparation of mouse tissuesSpleens were collected in PBS, pressed through a 70-μm cell strainer and washed with 10 ml of PBS with 1% FBS and 2 mM EDTA. Organs were then subjected to ACK lysis and passed through a 40-μm cell strainer to prepare the final single-cell suspension.
Interferon-γ (IFNγ) ELISpotThe IFN-γ ELISpot was performed according to manufacturer protocols (BD Biosciences). For each spleen and condition, 3 × 105 bulk splenocytes were plated in RPMI (supplemented with 10% FBS, 2 mM l-glutamine, penicillin and streptomycin) in duplicate wells and either incubated with (1) no antigen or stimulus; (2) 4.5 µg ml−1 protein antigen (mIgG, hIgG or TRACeR); or (3) phorbol myristate acetate (PMA)–ionomycin at working concentrations of 50 ng ml−1 and 1 µg ml−1, respectively. Cultures were incubated at 37 °C for 16 h and subsequently developed according to manufacturer protocols. IFNγ spots were imaged and counted on a CTL ImmunoSpot S6 universal analyzer.
Flow cytometryIn all cases of analysis of primary splenocytes ex vivo or following in vitro restimulation, we blocked nonspecific antibody binding using 10 µg ml−1 anti-CD16 and anti-CD32 (clone 2.4G2, BioXCell). For viability staining, we used both a live–dead fixable Aqua dead cell stain kit (1:500 dilution; Thermo Fisher Scientific) and a Zombie UV fixable viability kit (1:1,000 dilution; Biolegend). The following antibodies were used to stain primary cells: anti-CD44 BUV395 (clone IM7, 1 μg ml−1, BD Biosciences), anti-Ly-108 BUV737 (clone 13G3, 2 μg ml−1, BD Biosciences), antiCD127 BV421 (clone A7R34, 2 μg ml−1, Biolegend), anti-CD27 BV510 (clone LG.3A10, 2 μg ml−1, Biolegend), anti-CX3CR1 BV605 (clone SA011F11, 2 μg ml−1, Biolegend), anti-CD4 BV785 (clone RM4-5, 2 μg ml−1, Biolegend), anti-KLRG1 FITC (clone 2F1, 2 μg ml−1, Biolegend), anti-CD8 PE and PerCP-Cy5.5 (clone 53-6.7, 1 μg ml−1, Biolegend), anti-CD69 PE/Cy7 (clone H1.2F3, 2 μg ml−1, Biolegend), anti-CD3 AF700 (clone 17A2, 5 μg ml−1, Biolegend), anti-CD62L APC-eFluor 780 (clone MEL-14, 1.25 μg ml−1, Invitrogen), anti-Vβ 8.1, 8.2 TCR FITC and PE (clone MR5-2, 10 μg ml−1, BD Biosciences), anti-Vβ 8.3 TCR FITC and PE (clone 1B3.3, 10 μg ml−1, BD Biosciences), anti-CD107a BUV395 (clone 1D4B, 2 μg ml−1, BD Biosciences), anti-IFNγ BV605 (clone XMG1.2, 0.5 μg ml−1, Biolegend), anti-Ki-67 FITC (clone 11F6, 2 μg ml−1, Biolegend), anti-inteleukin 2 (IL-2) PE (clone JES6-5H4, 0.5 μg ml−1, Biolegend) and anti-tumor necrosis factor APC (clone MP6-XT22, 0.5 μg ml−1, Biolegend). For intracellular staining, we permeabilized the cells using the BD Cytofix/Cytoperm fixation/permeabilization kit according to the manufacturer’s protocols (BD Biosciences). To collect data, we used the CytoFLEX LX (Beckman Coulter) and BD LSRFortessa X-50; FlowJo 10.10 was used for analysis.
In vitro restimulation and intracellular stainingIn a 96-well U-bottom plate, 2 × 106 bulk splenocytes were plated and subsequently incubated with (1) no antigen; (2) 4.5 μg ml−1 protein antigen (mIgG, hIgG or TRACeR); or (3) PMA–ionomycin at working concentrations of 50 ng ml−1 and 1 μg ml−1, respectively. After 1 h of incubation at 37 °C, brefeldin A and monensin were added at final concentrations of 5 μg ml−1 and 2 μM, respectively. Additionally, we included BUV395 anti-mouse CD107a at a final concentration of 2 μg ml−1. Cultures were subsequently incubated at 37 °C for 10 h. After incubation, cells were subjected to surface and intracellular flow cytometric staining.
BiTE expression and purificationProtein expression of the \(\rm}_\;I,02}^--1}\) anti-CD3 fusion in the Drosophila melanogaster S2 cell line was performed as previously described for an analogous protein68. A DNA construct encoding the anti-CD3 fused with the \(\rm}_\;I,02}^--1}\) His-tagged at the C terminus was cloned in pMT vector and the S2 cells were stably transfected. The cultures were induced with 1 mM CuSO4 and, after 4 days, the supernatant was collected. The secreted protein was purified using a high-density metal affinity agarose resin (Agarose Bead Technologies) and was further purified by SEC using a HiLoad 16/600 Superdex 200-pg column at a flow rate of 1 ml min−1 in 150 mM NaCl and 20 mM sodium phosphate buffer (pH 7.4).
DLBCL cultureDLBCL cell lines were either directly received from the American Type Culture Collection (ATCC) or verified by ATCC short tandem repeat profiling. Cells were cultured at 37 °C in a 5% CO2 incubator with advanced RPMI (Gibco), 5% heat-inactivated FBS (Gibco), Glutamax (Gibco) and penicillin–streptomycin (Gibco). Cells were maintained between 200,000 and 1 million cells per ml and split every 2 days. Lines were routinely tested for Mycoplasma contamination using the universal Mycoplasma detection kit (ATCC).
Primary human T cell isolation and activationFor bispecific-induced cytotoxic assays, blood was collected from healthy, consenting donors by the Human Immunology Core at the University of Pennsylvania and total CD3+ T cells were enriched by magnetic separation. Cells were cultured in advanced RPMI (Gibco), 10% heat-inactivated FBS (Gibco), Glutamax (Gibco), penicillin–streptomycin (Gibco) and 10 mM HEPES (Quality Biological), supplemented with 300 U per ml recombinant IL-2 (National Cancer Institute (NCI) Biological Resources Branch (BRB)). T cells were cultured at 1 million cells per ml and activated with a 1:1 ratio of Dynabeads human T-activator CD3 and CD28 beads (Gibco) for 72 h, after which they were debeaded and expanded in IL-2-containing medium.
For generating CAR-T cells, primary CD8+ T cells were isolated from fresh leukopacks of anonymous donors (AllCells, LP, FR, 5B) by negative selection using the EasySep human CD8+ T cell isolation kit (STEMCELL Technologies, 17953). Isolated CD8+ T cells were cryopreserved in CELLBANKER 1 (AMSBIO, 11910). After thawing, T cells were cultured in human T cell medium consisting of X-VIVO 15 (Lonza, 04-418Q), 5% human AB serum and 10 mM neutralized N-acetyl l-cysteine (Sigma-Aldrich, A9165) supplemented with 30 U per ml of IL-2 (NCI BRB preclinical repository) for all experiments. After 24 h in culture, T cells were stimulated with human T-activator CD3 and CD28 Dynabeads (Life Technologies, 11131D) at a 1:1 ratio of cells to beads.
Bispecific-induced cytotoxicity assaysTumor cells were labeled with CellTrace CFSE (Invitrogen) at 1:1,000 in PBS for 15 min at 37 °C, following by extensive washout. Cells were counted and plated at 50,000 cells per well of a flat bottom 96-well plate. Primary human T cells were washed and 100,000 T cells were plated in each well. Bispecific antibody stocks were made by serial dilution and spiked into each well as indicated by dose curves. Where indicated, CD19–CD3-bispecific antibodies or CD19–βGal-bispecific antibodies were used as positive and negative controls (Invivogen). After ~18 h of coculture, Sytox blue (Invitrogen) viability stain was added to each well. Cells were analyzed by the high-throughput sampler of a LSR Fortessa (BD Biosciences). Tumors were gated from immune cells and the live fraction of each well was normalized to wells that received no antibody.
T cells incubated with off-target tumor cell lines SUDHL4 and SUDHL5 also displayed some activation in the presence of our TRACeR BiTE, albeit at a lower level (Supplementary Fig. 10). This is likely because of the activity of dimerized anti-CD3 scFv, which can facilitate crosslinking of CD3 on T cells and facilitate T cell activation independent of target recognition and killing.
X-ray crystallography and structure determinationThe HLA-A*2:01/NY-ESO-1–MAM–TRACeR complex was prepared by mixing MAM–TRACeR and HLA-A*2:01/NY-ESO-1 at a 1:1.3 molar ratio; the mixture was incubated at 4 °C for 1 h. Unbound proteins were separated by SEC using a Superdex200 increase 10/300 GL column, run at 0.5 mg min−1 flow rate with running buffer, 25 mM Tris pH 8.0 and 150 mM NaCl. The complex was concentrated to 10.4 mg ml−1 for crystallography and the sample was mixed at a 1:1 ratio with reservoir well solution and incubated at 20 °C. Crystals were obtained in 200 mM sodium sulfate and 20% w/v PEG 3350 using the sitting drop vapor diffusion method. Crystals were harvested after transferring to cryogenic solution containing reservoir solution and 20% v/v glycerol. Crystals were screened and data were collected on the National Synchrotron Light Source (FMX, 17-ID-2) at Brookhaven National Laboratory (BNL) using a Dectris Eiger 16M detector. Data were processed using XDS69 and the structure of HLA-A*02:01/NY-ESO-1–MAM–TRACeR complex was solved by molecular replacement method using the Molrep70 and REFMAC 5 (ref. 71) software of the CCP4 package suite72. The structure of HLA-A*02:01/NY-ESO-1 (PDB 1S9W)35 and a structural model of MAM–TRACeR were used as search models. The complex structure was refined using BUSTER (Global Phasing) and PHENIX73 and Coot74 were used for model building.
CAR-T cell cytotoxicity assaysCD8+ Primary human T cells expressing constitutive \(\rm}_\;I,02}^--1}\) CAR were counted and stained with CellTrace yellow dye (Invitrogen, C34567) at a concentration of 2.5 μM while otherwise following the manufacturer’s protocol. Similarly, DLBCL tumor cell lines were counted and stained with CellTrace violet dye (Thermo Fisher Scientific, C34571) at a concentration of 2.5 μM. After staining, cells were counted, resuspended in their respective media and adjusted to a concentration of 2 × 105 cells per ml for T cells and 105 cells per ml for each DLBCL tumor cell line. Then, 100 μl of T cells and a 100 μl of target cells were combined in a in a 96-well round bottom plate, for an effector-to-target ratio of 2. T cells and DLBCL tumor targets were cocultured at 37 °C with 5% CO2. After 18 h, cultures were analyzed for specific lysis of target tumor cells by flow cytometry. All cells were stained with live–dead fixable near-infrared dead cell stain kit at 1:500 dilution (Invitrogen, L10119) and human anti-CD8 FITC antibody (1:100 dilution; BD Biosciences, 347313) for 30 min in ice. After washing in flow buffer, cells were resuspended and acquired in a NovoCyte Quanteon flow cytometer (Agilent). After cells were gated by size and doublets were excluded, only live events were considered. T cells were differentiated of target cells by CellTrace and CD8 expression. The percentage of specific lysis of target cells was determined by comparing the number of target cells alive in the coculture with TRACeR CAR to the average number of target cells alive in the coculture with untransduced T cells. All flow cytometry data analysis was performed in FlowJo X software (TreeStar) and GraphPad Prism 9 (Dotmatic). The following equation was used to calculate the percentage of specific lysis:
$$\begin\% \,specific\,lysis\\=\left(1-\frac\right)\times 100\end$$
Sequence conservation analysisAmerican HLA haplotype (allelic) frequencies were obtained from the BeTheMatch registry75 across five broad race groups (AFA, API, etc.) and converted to diploid (population) frequencies assuming a Hardy–Weinberg equilibrium. A combined American population frequency for each allele was obtained by normalizing values according to the racial distribution followed by the 2020 US Census. ‘Common’ alleles were defined as those with a haploid frequency greater than 0.05% in at least one of the five broad race groups for a total of 215 HLA alleles across HLA-A, HLA-B, and HLA-C loci. HLA allele sequences were obtained from the IMGT/HLA database22 and a pairwise sequence alignment was performed on the HLA-A*02:01 heavy chain sequence (consisting of 180 residues from the N terminus). Relevant HLA contact residues were identified by visual inspection and a sequence logo was created using the logomaker (version 0.8)76 Python package. Visualizations were produced with PyMOL (version 2.5.2).
TRACeR–pMHC I structural modelingFor structural modeling, all target TRACeR sequences were threaded onto the original TRACeR–NY-ESO-1 crystal structure. Then, the target TRACeR model along with the target pHLA-I complex were superimposed onto the original TRACeR–NY-ESO-1 crystal structure on the basis of Ca atoms and the original TRACeR–pHLA complex was deleted using PyMOL (version 2.5.2). Finally, the modeled complex was optimized using the FastRelax protocol77 in Rosetta78 (version 2020.08) and the relaxed model with the lowest total ref2015 score was selected as the final TRACeR–pHLA model.
The pHLA-I complexes were obtained as follows. For Nef, the highest-resolution pHLA-I crystal structure was identified from HLA3DB (ref. 20) as PDB 3VXN (ref. 79). For KRAS-G12V, the HLA-A*11:01 sequence was threaded onto the Fab-bound KRAS-G12V/A*03:01 complex (PDB 7STF (ref. 60)) using the RosettaRemodel38 application from the Rosetta (version 2020.08)4 suite of programs and the Fab and β2m were removed. For PRAME and SARS-CoV-2, the pMHC I complex was modeled using RepPred, a state-of-the-art homology modeling method20.
Ethics statementThe animal studies received approval from the IACUC of the Office of Animal Welfare at the University of Pennsylvania (protocol 803042). Blood-related research was conducted under biosafety project approval by the Environmental Health and Safety Office at Stanford and under approval from the University of Pennsylvania review board. Written informed consent in this study was provided by all participants.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)