
記住我






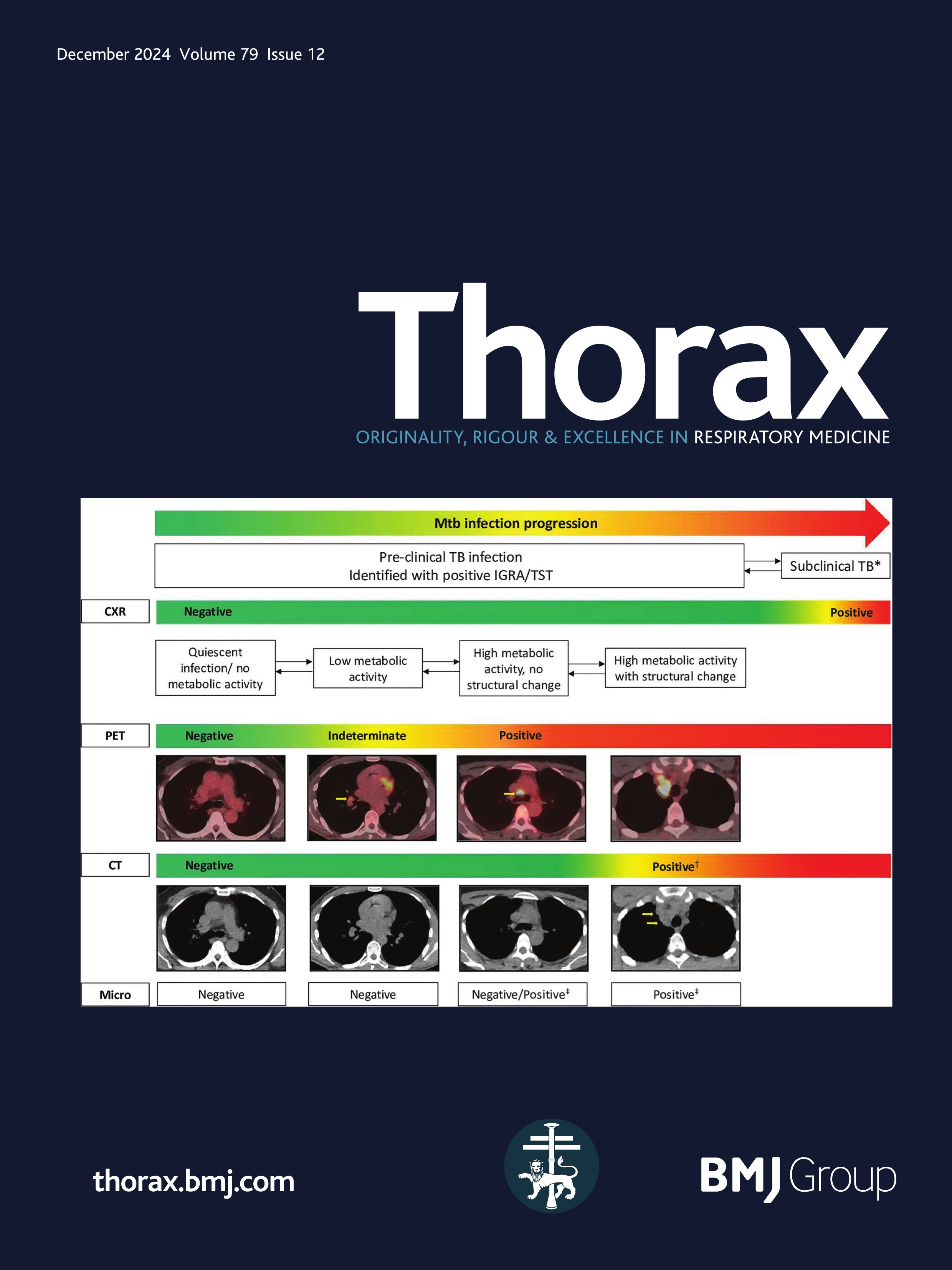
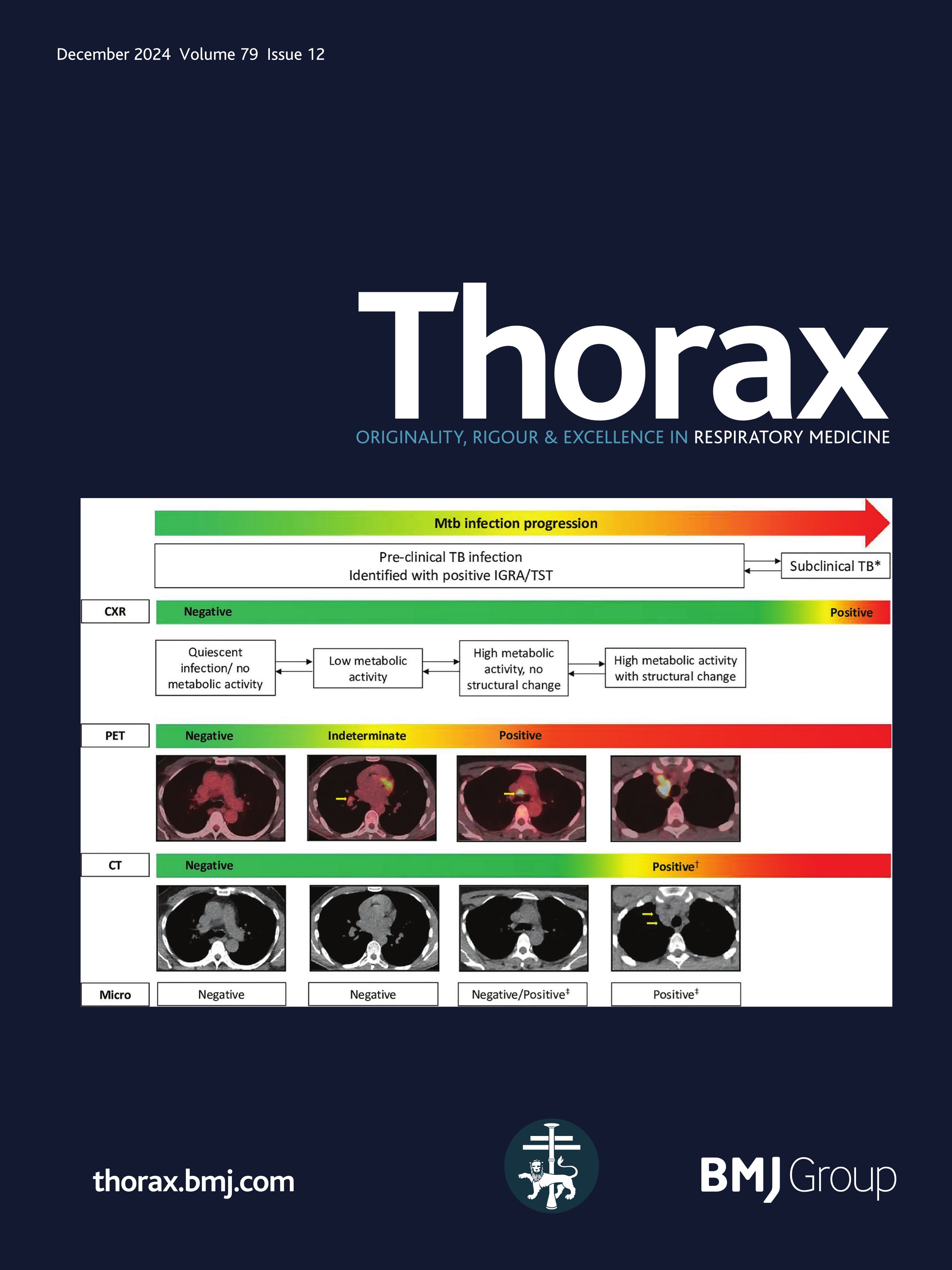
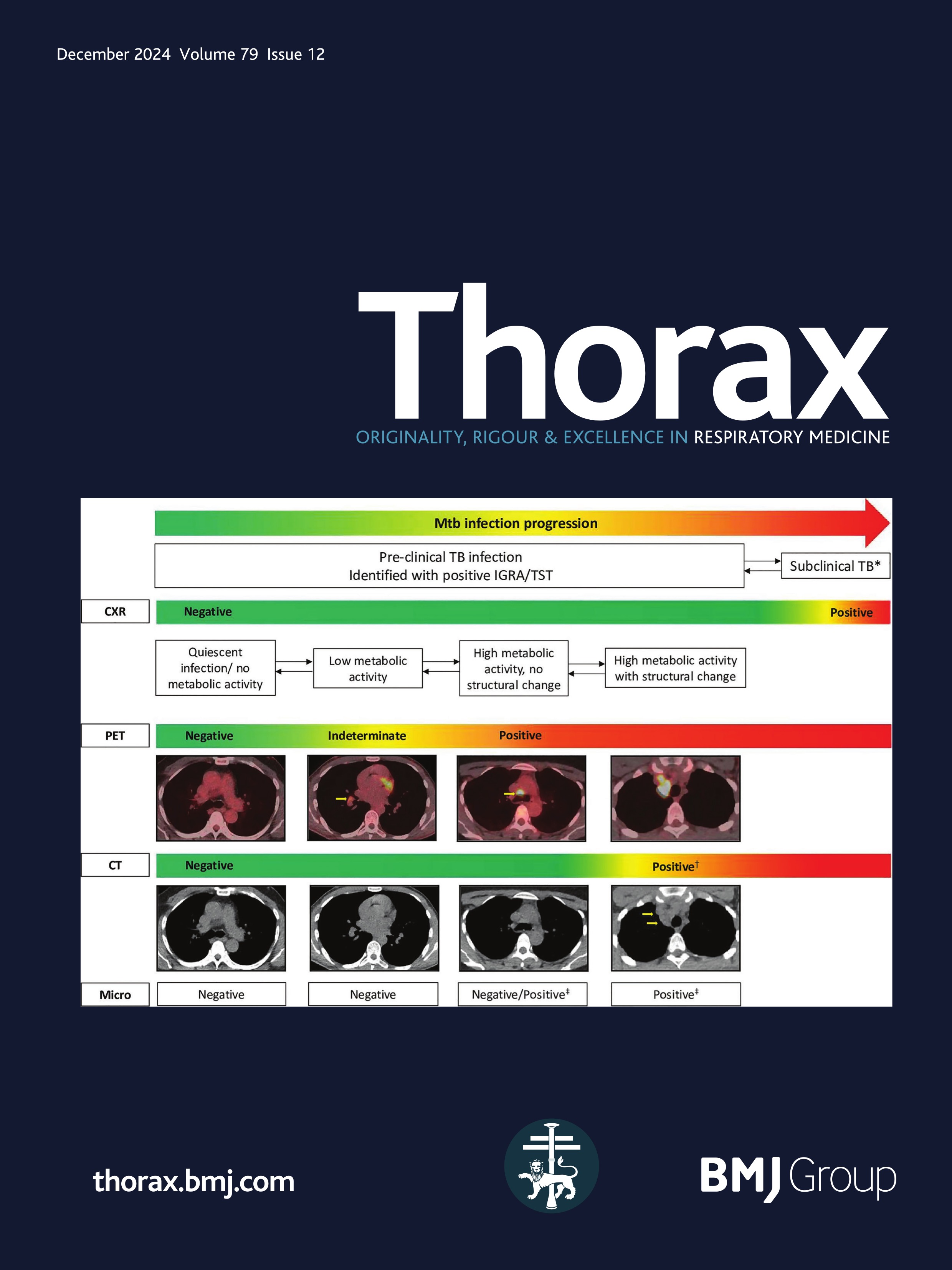

Childhood interstitial lung disease (chILD) comprises a diverse group of rare respiratory disorders affecting infants, children and adolescents, some of which result in pulmonary fibrosis.1–3 Mortality in chILD cohorts may be substantial and estimates have varied in prospective studies between less than 1%4 and 30% among infants and 13% overall.5 The outcome of individual disease entities is largely unknown, and the exact causes of death often remain unclear. In adults, pulmonary fibrosis is a major cause of death in patients with ILD.6 In contrast, in paediatric patients, pulmonary fibrosis is a rare event, with few reports and no systematic prospective studies.7 In adults, the definition of fibrosis, early signs of fibrosis and features of progressive pulmonary fibrosis have been extensively investigated, discussed and agreed on in international consensus statements of respiratory societies.8–11 In paediatric patients, however, no diagnostic criteria have been established for pulmonary fibrosis and other aspects of fibrotic disease behaviour. Several chILD disorders are genetic in origin, with different defects (and phenotypes) manifesting with increasing age throughout life.12 While most children with severe lung developmental conditions such as acinar dysplasia or alveolar capillary dysplasia die in the neonatal period or during infancy,13 14 it has become clear that many children with chILD can survive into adulthood, where their disease will fall under the remit of adult physicians and pulmonologists. There is an unmet need for bridging chILD to adult ILD. The goal of this article is to provide state-of-the-art information on fibrosing/potentially fibrosing lung disease in childhood and highlight issues to be addressed in future studies.
Definitions of fibrosis in childrenFibrosis is defined histopathologically as an increase in collagen deposition and fibroblasts, which may lead to loss of gas-exchange tissue accompanied by lobular remodelling of the parenchyma and microscopic honeycombing (figure 1). The lung can respond to injury with different patterns of interstitial fibrosis, including fibrosing non-specific interstitial pneumonia (NSIP), and, though rare in childhood, usual interstitial pneumonia. The pathology of fibrosing ILD in children overlaps with that in adults, although lobular remodelling with smooth muscle hyperplasia is more prominent in young children (table 1).
 Figure 1
Figure 1 Findings of paediatric pulmonary fibrosis on lung histology. (A) Fibrotic NSIP with uniform expansion of alveolar septa by collagen (arrows) in a 20-month-old with an SFTPC mutation (H&E). (B) UIP pattern in an adolescent with an ABCA3 mutation consists of patchy dense fibrosis accentuated in the subpleural space with honeycomb cystic remodelling (*enlarged, irregular airspaces, H&E) and fibroblastic foci (insert (see arrow heads), Movat stain).(C) Focal dense periairway fibrosis (arrow) is present in a child with restrictive lung disease after bone marrow transplant (Movat stain). (D) Lobular remodelling in a child with NKX2.1 deficiency is characterised by cystic bronchiolar metaplasia (*) interspersed by bands of smooth muscle (arrows; H&E). ABCA3, adenosine triphosphate-binding cassette subfamily A member 3; NSIP, non-specific interstitial pneumonia; NKX2.1, NK2 homeobox 1; SFTPC, surfactant protein C; UIP, usual interstitial pneumonia.
Table 1Criteria for the definition of fibrosing ILD
While the resolution of CT imaging is much less than that of a lung biopsy, it allows for the assessment of the entire lung parenchyma for findings suggestive of lung disease and fibrosis. Definite signs of fibrosis on CT in children and adults include evidence of traction bronchiectasis and bronchioloectasis (figure 2A) and honeycombing (figure 2B). Architectural distortion is characterised by displacement of pulmonary vessels, airways, fissures and septa (figure 2C and table 1). Areas of reticular opacites, often sub-pleural, are further evidence for fibrosis (figure 2A and table 1). Several of these findings, and especially reticular opacities, can be observed with or without associated ground glass opacities. Importantly, cystic lucencies, particularly when accompanied by ground glass opacities, are an early characteristic of genetically defined surfactant dysfunction disorders (figure 2D), which frequently manifest with fibrosis early in life15 16 (see disease-specific sections below). Often the walls of these cysts are barely visible, due to the lack of a thicker fibrous or epithelial wall that often defines cysts.17 Based on current data, these cystic lesions are included in the imaging criteria for fibrosis. Correlation of these cystic lesions in chILD to the histopathology is needed.
 Figure 2
Figure 2 Findings of paediatric pulmonary fibrosis on chest CT. (A) Reticular opacity consisting of mesh/net-like opacities associated with traction bronchiectasis with irregular bronchiolar dilation (yellow arrows) in an 11-year-old with a history of ARDS. (B) Honeycombing consisting of clustered enlarged airspaces with defined walls in a 15-year-old male with chILD of unknown aetiology. (C) Architectural distortion with deformation of the fissures, parenchymal bands consisting of irregular linear opacities, some of which extend to the pleural surface (yellow arrows) and traction bronchiectasis (red arrow) in a 19-year-old female with post-COVID-19 fibrosis. (D) Cystic lucencies with rounded areas of decreased density without defined wall (yellow arrows) in a 10-year-old patient with an ABCA3 mutation. ABCA3, adenosine triphosphate-binding cassette subfamily A member 3; ARDS, acute respiratory distress syndrome; chILD, childhood interstitial lung disease.
Imaging findings in children need to be correlated with patients’ clinical presentation and history and differentiated from lung abnormalities identified on CT scans obtained for other purposes, including staging scans for malignancies, or patients with known cardiac or lung disease not associated with fibrosis. Such findings have been labelled as interstitial lung abnormalities in adults and are present in 33% of undiagnosed first-degree relatives of patients with IPF,18 although they have not yet been investigated in children.
Understanding that lungs are constantly developing in children and that the postnatal lung has an enormous repair potential into adolescence is of pivotal importance when considering the genesis of fibrosis in childhood. How fibrosis evolves in the developing lung, and during the disease course of various conditions, and if all aspects are irreversible, is largely unexplored and requires investigation.
Definition of progressive fibrosis in childrenIn adults, progressive pulmonary fibrosis is defined in clinical practice guidelines by worsening of any two of the following three parameters over 12 months with no alternative explanation: respiratory symptoms, lung function (absolute decline in forced vital capacity ≥5% or absolute decline in diffusing capacity of the lung for carbon monoxide (DLCO) ≥10%) or radiological changes.10 ‘Radiological changes’ encompass increased extent or severity of traction bronchioloectasis and bronchiectasis, new associated ground-glass opacity, new fine reticulation, increased extent or coarseness of reticulation, new or increased honeycombing and increased volume loss.10 Longitudinal data in children are scarce, but cystic lesions increase over time in number and size in both surfactant protein C (SP-C) deficiency15 and adenosine triphosphate-binding cassette subfamily A member 3 (ABCA3) deficiency,16 again supporting their role in paediatric lung fibrosis.
ILD disease trajectory may differ between adults and children of different age groups2 with possible differences in histological and radiological findings between children and adults. Changes in lung function in children occurring in the context of ongoing postnatal lung development further complicate the identification of progressive fibrosis in children.7 In addition, in children, valid lung function measurements are usually not available until the age of 6 years and DLCO measurement is not available until the vital capacity is at least 1000 mL. Thus, detection of disease progression in young children with chILD is difficult, and not possible using adult criteria.
chILD natural disease course and pulmonary fibrosisIn the last decade, the increased availability of genetic testing has led to the discovery of many genetic defects associated with novel entities of chILD, with different phenotypes and pathologies manifesting with increasing age.12 However, the natural history of most chILD entities is not yet determined. Whereas some remit and never become symptomatic in adults,19 20 chILD disorders may also have sustained chronic parenchymal inflammation and progress to fibrosis.21 22 In adults, data on the exact or estimated proportion of patients with progressive fibrosis among the various types of ILD are limited23 while in children no such data are available. As an initial approach we searched the European- and US-chILD registries for the presence or absence of cases with diagnosed fibrosis.
Fibrosis in different chILD entitiesAnalysis of the chILD-EU and US-chILD registries identified 47 distinct entities where fibrosis has been recorded (box 1).
Important entities manifesting fibrosis are discussed below. Figure 3 shows lung histology in some of these entities while CT patterns are shown in figure 4.
 Figure 3
Figure 3 Lung histology findings of children with specific chILD entities. (A) Cellular and fibrotic NSIP seen in an adolescent later diagnosed with polymyositis (H&E).(B) Child with SAVI has dense interstitial fibrosis and occlusive airway fibrosis (Movat stain).(C) Peribronchiolar fibrosis with isolated multinucleated giant cells (arrows) in a child with hypersensitivity pneumonitis (H&E). chILD, childhood interstitial lung disease; NSIP, non-specific interstitial pneumonia; SAVI, stimulator of interferon genes-associated vasculopathy of infancy.
 Figure 4
Figure 4 CT patterns of children with specific chILD entities. (A) SFTPC mutation in a 6-year-old with reticular opacities and cystic lucencies (yellow arrows) on axial CT image. (B) SAVI in a patient aged 7 years, with diffuse ground glass and reticular opacities with large cystic lesions on axial CT image. (C) COPA syndrome in a 13-year-old with diffuse reticular opacities and cystic lucencies (yellow arrows). (D, E) PPFE in a 15-year-old with papillary thyroid cancer with pleural thickening (red arrows), septal thickening, yellow arrows, traction bronchiectasis (green arrows) and pneumothorax (blue arrows) on (D) axial and (E) coronal images. COPA, coatomer protein subunit alpha; SAVI, stimulator of interferon genes-associated vasculopathy of infancy, SFTPC, surfactant protein C gene.
Box 1 Conditions in the chILD-EU and US-chILD registries where pulmonary fibrosis has been described, ordered according to their disease categories (lung-native disorders (native diffuse parenchymal disorders), systemic disease-related disorders and exposure-related disorders).Lung-native disorders (lung only)Surfactant protein C mutations
ABCA3 deficiency
Non-specific interstitial pneumonia
Combined pulmonary fibrosis and emphysema
Bronchopulmonary dysplasia
Bronchiolitis obliterans
Surfactant protein B mutations
Wilson Mikity, new bronchopulmonary dysplasia
Systemic disease-related disordersSTING-associated vasculopathy (SAVI)
Hermansky-Pudlak Type 2
FARS1 associated disease
Juvenile systemic sclerosis
Mixed connective tissue disease
Undifferentiated connective tissue disease
Collagen vascular disorders
GATA2
Systemic juvenile arthritis-associated lung disease
Sarcoidosis
Bone marrow transplantation
Bronchiolitis obliterans after bone marrow transplantation
RAB5B deficiency
COPA defect
Systemic lupus erythematosus
STAT3 GOF mutation
TBX4 mutation
NKX2.1 gene defect
MARS-associated disease
Telomerase complex mutations
Integrin α3-disease
MDA-5 jDM
Hereditary PAP
Rubinstein-Taybi syndrome
FINCA syndrome
Juvenile myelomonocytic leukaemia
Antisynthetase syndrome
IgG4-related disease
Ectodermal dysplasia
Ataxia telaniectasia
Related to trisomy 21
Filamin A mutation
FGF10
Granulomatosis with polyangitis
Exposure-related disordersRadiation lung injury
Hypersensitivity pneumonitis
Drug reactions
Toxic inhalation
Aspiration syndromes
ABCA3, adenosine triphosphate-binding cassette subfamily A member 3; chILD, childhood interstitial lung disease; COPA, coatomer protein subunit alpha; FARS1, phenylalanine-tRNA synthetase 1; FINCA, fibrosis, neurodegeneration and cerebral angiomatosis; GATA2, GATA binding protein 2; MARS, methionyl-tRNA synthetase; NKX2.1, NK2 homeobox 1; PAP, pulmonary alveolar proteinosis; RAB5B, Ras-related protein Rab-5B; STAT3-GOF, signal transducer and activator of transcription 3 gain of function; STING, stimulator of interferon genes; TBX4, T-box transcription factor 4.
Conditions discussed further below are in bold.
Lung-native parenchymal disordersSurfactant dysfunction disorders caused by pathogenic variations in the genes for ABCA3 (ABCA3) and SP-C (SFTPC) can manifest in childhood with consequences in later life.15 24
ABCA3 deficiencyABCA3 is an intracellular transmembrane glycoprotein that facilitates the packaging and transport of surfactant into lamellar bodies of type 2 pneumocytes.25 More than 200 ABCA3 variants leading to loss of function have been described and are implicated in inherited pulmonary diseases with varying phenotypes, from potentially fatal neonatal respiratory distress syndrome to diffuse parenchymal lung diseases in childhood or even adulthood.16 24 26 Progression to fibrosis is frequently reported in patients with ABCA3 mutations and ILD, generally increasing with age.16 24 27 Figures 1B, 2D show lung histological and chest CT characteristics of children with ABCA3 deficiency.
SP-C dysfunction disorderSP-C dysfunction disorder is an inherited autosomal dominant disease caused by a mutation in the SFTPC gene.15 28 29 A former study categorised 17 patients who had a heterozygous dominant mutation in the SFTPC gene according to their typical characteristics and age.15 Three categories of presentation were defined as (1) neonatal respiratory distress syndrome, as originally described for surfactant protein B deficiency,30 often with a family history of lung disease, previous pregnancy, miscarriage or postnatal death; (2) the classical presentation of infants with tachypnoea, dyspnoea and failure to thrive, diffuse ground glass opacity on chest CT within the first few months, with pathological findings of NSIP, or a combination of pulmonary alveolar proteinosis (PAP), NSIP and desquamative interstitial pneumonia and (3) late childhood, adolescence or adulthood diagnosis of chronic pulmonary fibrosis15 31 with progressive findings of increased cystic lucencies and reticular opacities on chest CT.32 Figures 1A, 4A show lung histological and chest CT characteristics of children with an SFTPC mutation.
Systemic disease-related disordersChildren often present with pulmonary manifestations of systemic diseases, which can be associated with high morbidity and mortality. In a subset of patients, pulmonary disease may be the predominant initial clinical presentation. It is, therefore, important for paediatric pulmonologists to consider systemic diseases when making a diagnosis.33
Systemic disease-related disorders in immunocompetent childrenConditions with a predominant lung phenotypeCauses of chILD that have been linked with pulmonary fibrosis include mutations affecting fibroblast growth factor 10 (FGF10) and its receptor FGFR2,34 brain–thyroid–lung syndrome/NKX2.1 deficiency,35 T-box transcription factor 4 (TBX4) syndrome,36 filamin A (FLNA) deficiency37 38 and RAB5 deficiency.39 Due to the rarity of these diseases, their frequency and the extent of fibrosis are largely unknown.
MARS1 deficiencyA severe form of childhood PAP caused by biallelic missense mutations in the methionyl-tRNA synthetase 1 gene (MARS1) has been reported, with high prevalence localised to the island of La Réunion in the Western Indian Ocean.40 MARS1 encodes for cytosolic MARS1 and plays a pivotal role in protein biosynthesis. Patients present with respiratory insufficiency, PAP, progressive pulmonary fibrosis (in about 50% by the age of 14 years), and structural hepatopathy, with high rates of mortality reported.41 A recent study of two cases with pathogenic biallelic MARS1 gene mutations demonstrated a multisystemic phenotype not restricted to the lung and liver, treatable with a methionine-rich diet.40 CT findings in patients with MARS1 deficiency have included ground glass opacities with septal thickening resulting in a crazy paving pattern.40 42
FARS1-related disordersThere have been a small but growing number of reports on phenylalanine-tRNA synthetase (FARS)1-related disorders. FARS1 is part of the ubiquitously expressed family of aminoacyl-tRNA synthetases (ARS), which play an essential role in protein biosynthesis and other functions outside of translation.43 Biallelic mutations of each of the two FARS1 subunits—FARSA and FARSB—have been reported, with FARS-beta-related disorders being slightly more common.44 A distinct pulmonological phenotype has been described as childhood-onset chronic ILD characterised by bilateral ground glass opacities on CT, histological cholesterol pneumonitis and PAP.44 Additional organ involvement includes intracranial aneurysms, glomerulosclerosis, failure to thrive, cerebral calcifications, hypotonia, immune dysregulation and liver cirrhosis.44–46
Fibrosis, neurodegeneration and cerebral angiomatosis syndromeRare biallelic missense variants in NHL repeat containing protein 2 (NHLRC2) have recently been discovered to cause fibrosis, neurodegeneration and cerebral angiomatosis (FINCA) syndrome, a combination of pulmonary fibrosis, neurodegeneration and cerebral angiomatosis.47 The associated phenotype can be lethal in infancy, and a small number of cases have reported initial presentation of ILD combined with neurodevelopmental delay and multiorgan involvement.47 48 Recently, a study using the chILD-EU register database found that some patients experience stabilisation of their lung disease over time and survive into childhood, suggesting that FINCA syndrome should be considered at all ages, including in patients with a history of stabilising chILD who have neurodevelopmental delay.49 Chest CT findings were variable including ground glass opacities, consolidation, cysts and overinflation. Of interest, NHLRC2 expression is increased in idiopathic pulmonary fibrosis in adults.50
Systemic disease-related disorders with features of immunodeficiency/Immunodysregulation/autoinflammationRheumatological disordersPulmonary disease is more common in certain autoimmune rheumatological diseases typically diagnosed after 6 years of age and driven by the adaptive immune system (ANCA-associated vasculitis, Ribonucleoprotein (RNP)-associated systemic lupus erythematosus (SLE), melanoma differentiation-associated protein 5 (MDA-5) juvenile dermatomyositis (JDM), juvenile systemic sclerosis (jSSc).
ANCA-associated vasculitisIn paediatric ANCA-associated vasculitis (pAAV), pulmonary involvement is very common, with higher prevalence in eosinophilic granulomatosis with polyangiitis (EGPA (88%)) compared with GPA (52%–74%) and microscopic polyangiitis (MPA (29%–44%)) although EGPA is much less common51; often pAAV presents with periods of high acuity with overt respiratory symptoms such as chronic cough, exertional dyspnoea, haemoptysis and respiratory distress secondary to haemorrhage and vasculitis, especially in GPA compared with MPA and unclassified AAV. However, paediatric patients can also be asymptomatic with their lung disease evolving into fibrotic interstitial changes,52 especially in MPA. Imaging findings of AAV include ground glass opacities and areas of consolidation as well as centrilobular and larger nodules, characteristic of pulmonary haemorrhage.53
Systemic lupus erythematosusSLE has a high prevalence of pulmonary disease (up to 40% within the first year)54 ; a common acute presentation with a good prognosis is pleuritis; less common acute presentations involve acute lupus pneumonitis (ALP; <10%) of which about 3% may lead to chronic ILD.55 The increased risk factors for ILD (eg, anti-RNP, ANCA, severe SLE activity) remain controversial. Common findings on CT in patients with SLE include ground glass opacities, septal thickening and diffuse consolidation, each of which are seen in more than 50% of patients. Other common findings include patchy consolidations, fibrotic changes and pleural effusions.56 Other chronic presentations include shrinking lung disease (<1%) and pulmonary hypertension (5%–14%).
Melanoma differentiation-associated protein 5 juvenile dermatomyositisMDA-5 associated JDM (<5% of total JDM subtypes in the USA and UK) has a high prevalence of rapidly progressive ILD leading to alveolar fibrosis.57 In a Japanese study, 31% of patients with MDA-5 JDM developed ILD, which was rapidly progressing in 50% of cases.58 Radiological patterns in MDA-5 JDM are often described when the patient is symptomatic with a rapidly evolving course with ground glass opacities and multifocal areas of consolidation, sometimes complicated by pneumothorax and pneumomediastinum. In some cases, this may progress quickly to findings of fibrosis including reticular opacities, traction bronchiectasis and architectural distortion.59
Juvenile systemic sclerosisjSSc is an autoimmune disease with an indolent subclinical pulmonary course that can be detected early. Although jSSc is rare, pulmonary disease ranges from 35% to 55% and includes ILD, pulmonary arterial hypertension secondary to ILD and vasculopathy.60 Early chest CT findings typically reflect alveolitis, with ground glass opacities and micronodules that progress slowly to fibrosis with findings of reticular opacities, traction bronchiectasis, honeycombing, cystic changes and architectural distortion.61 Despite these findings, many patients with jSSc can be asymptomatic, especially early in the disease course.
Autoinflammatory diseasesUnlike autoimmune rheumatological disease, autoinflammatory diseases develop earlier in childhood and are driven more by the innate immune system. These include COPA (coatomer protein complex subunit alpha) (syndrome) and stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI).
Coatomer protein complex subunit alphaCOPA is a monogenic endoplasmic reticulum stress disease leading to lung inflammation and scarring typically presenting (76%) before age of 5 years62; pulmonary manifestations (chronic cough, tachypnoea, dyspnoea) are common due to capillaritis and alveolar haemorrhage or ILD associated with lymphoid hyperplasia and follicular bronchiolitis, lymphocytic interstitial pneumonia (LIP), pulmonary cysts and fibrosis; a unique CT pattern is diffuse ground glass opacity, septal thickening and cysts and centrilobular nodules. Common chest CT findings such as ground glass opacities and pulmonary nodules, can improve while cysts, septal thickening and fibrosis may progress (figure 4C).63 As COPA has variable clinical penetrance, patients can be asymptomatic but universally develop progressive lung disease with worsening pulmonary function due to pulmonary haemorrhage, which can be insidious, or ILD.
STING-associated vasculopathy with onset in infancySAVI is a monogenic, autosomal dominant type I interferonopathy caused by gain-of-function variants in transmembrane protein 173 (TMEM173), which presents typically early in life with cough, tachypnoea, exertional dyspnoea, haemoptysis in association with ILD (75%). Chest CT imaging demonstrates ground glass opacities, septal thickening, reticular opacities and cysts that can be seen in lung fibrosis.64 However, the severity and natural course of lung disease vary even within the same mutation.65 Figures 3B, 4B show lung histological and chest CT characteristics of children with SAVI.
Hermansky-Pudlak syndromeHermansky-Pudlak syndrome (HPS) is a rare hereditary disorder of albinism and bleeding diathesis.66 There are at least 11 different forms of HPS caused by mutations in different genes.66 67 While all have oculocutaneous albinism and bleeding diathesis due to platelet dysfunction, other organ manifestations are variable. For HPS1 and HPS4, pulmonary fibrosis has been reported in early adulthood but typically occurs in middle-aged adults,68 69 whereas children with HPS2 may develop ILD and pulmonary fibrosis.66 In the lungs, dysfunctional type 2 epithelial cells interact with macrophages, other immune cells and fibroblasts to generate fibrotic remodelling of the tissue.66 70 71 CT findings in patients with HPS include ground glass opacities, reticulation and traction bronchiectasis.
STAT3-GOF variantsSignal transducer and activator of transcription 3 gain-of-function (STAT3-GOF) variants cause severe multisystem autoimmunity, with ILD manifestations developing in approximately one-third of patients. A recent case study identified two new lung-predominant STAT3 variants, which manifested as severe ILD during the first 3 years, highlighting the importance of closely monitoring pulmonary function in patients known to carry STAT3-GOF alleles.72 Other disorders associated with immune-dysregulated ILD in children include LIP and granulomatous lymphocytic ILD.
TelomeropathiesWhile mutations in genes of the telomere complex can play a major role in fibrosis in adults,31 this is relatively rare in paediatric patients but may cause fibrosis.73 In children with variants of the dyskeratosis congenita gene, DKC1, lung fibrosis can also be observed (Hoyeraal-Hreidarsson syndrome).74
Exposure-related disordersExogenous allergic alveolitis/hypersensitivity pneumonitisHypersensitivity pneumonitis (HP) is a group of immunologically mediated lung diseases caused by an immunoglobulin G-mediated hypersensitivity reaction to an environmental trigger. This leads to inflammation of the distal bronchioles and alveoli, often with granulomas and, if not diagnosed and treated, may lead to chronic severe lung disease including progressive fibrosis, emphysema and secondary pulmonary hypertension.75 Figure 3C shows lung histological fibrosis characteristics of a child with HP. CT findings of HP include ground glass opacities, centrilobular nodules and mosaic attenuation in the acute and subacute phases and can progress to include fibrotic changes such as reticulation, traction bronchiectasis and honeycombing in chronic HP.
Pulmonary fibrosis or pleuroparenchymal fibroelastosis following radiation, chemotherapy or stem cell transplantIt has long been recognised that allogenic bone marrow transplantation in children is associated with fibrotic pulmonary complications.76 In the last decade, there have been developments in our understanding of ILD entities following autologous stem cell transplant, including non-infectious idiopathic pneumonia syndrome (IPS).77 More recently, in a study of 340 paediatric patients treated with high-dose chemotherapy and autologous haematopoietic stem cell transplantation (HSCT), 2.4% developed non-infectious ILD.78 Lung biopsies showed interstitial fibrosis and/or septal expansion from inflammatory cells and pleural thickening.78
Pleuroparenchymal fibroelastosis (PPFE) represents fibrosis of the pleura and subpleural parenchyma, dominating the upper lung lobes. On CT, patients present with areas of pleural thickening, traction bronchiectasis, architectural distortion and consecutive volume loss (figures 4D, E). Many patients may have a flattened chest (platythorax). Pneumothorax is a common complication. Histology shows fibrosis with a particular abundance of elastic fibres. In contrast to adults, in children only one case of idiopathic PPFE has been reported,79 with secondary PPFE more frequent, having an incidence of 4% after HSCT and previous chemotherapy or radiotherapy.80 Following SCT, the median overall survival with PPFE was only 6.8 years.80 Whereas the combined presence of bronchiolitis obliterans, organising pneumonia and IPS increased the risk of PPFE by about 2.5-fold, pneumonia more than 3 months after SCT increased the risk by about 10-fold. In lung transplanted subjects, PPFE may be a manifestation of chronic lung rejection.81
Treatment of fibrosing chILDPulmonary fibrosis is a consistent response pattern of the lungs to a variety of injuries. As indicated above, these include endogenous factors like autoinflammatory, autoimmune and strong genetic drivers, and noxious exogenous factors. Second hits, such as acute exacerbations due to respiratory tract infections, have been shown to have a significant and deleterious effect on the clinical course of and health-related quality of life in chILD.82 Thus, management targeting these underlying factors is likely to ameliorate the driving forces towards inflammation and fibrosis.7
All recommendations are limited by the absence of appropriate randomised controlled clinical trials. Besides supportive treatments in the form of oxygen supplementation and/or ventilation, adequate caloric intake, psychosocial support and physiotherapeutic rehabilitation, clinical management is currently guided by indirect evidence, case reports and clinical experience.1 83–86 Prophylactic immunisations may reduce the risk of exacerbations.87 Patients with autoimmune disorders may be treated with steroids1 and potentially steroid-sparing immunosuppressive medications such as mycophenolate mofetil (MMF), azathioprine, intravenous immunoglobulin, cyclophosphamide and calcineurin inhibitors.88 Inhibitors of Janus kinase such as baricitinib and ruxolitinib may also be an effective treatment of autoinflammatory interferonopathies, such as SAVI, STAT3-GOF and COPA, and several case studies have demonstrated promising results.89–91 In addition, biologics such as rituximab, an anti-CD20 monoclonal antibody, have shown promising effects in treating immune-mediated diseases, with several studies showing efficacy in treating systemic sclerosis in adults and children, including one study combining rituximab with MMF.92–95
The anti-inflammatory agents hydroxychloroquine and azithromycin have been included in treatment protocols for chILD.1 The first randomised, placebo-controlled trial in chILD, which included a large fraction of potentially fibrosing chILD with a mean forced vital capacity of approximately 50% of predicted, was a recently completed phase II study of hydroxychloroquine in 29 children.96 Although the study failed to document treatment efficacy, partly because of its small size, hydroxychloroquine was well tolerated. While larger studies will be necessary, the results suggest that the previous optimistic appraisal of hydroxychloroquine use in chILD should be evaluated further.
The InPedILD trial, a double-blind, randomised, placebo-controlled clinical trial of nintedanib in children and adolescents (6–17 years) with clinically significant fibrosing chILD, used a definition of paediatric lung fibrosis based on the imaging and histology criteria to identify eligible patients for enrolment.97 Fibrosis was diagnosed by the presence of one of the biopsy criteria and one or more high-resolution CT (HRCT) findings, excluding cystic abnormalities, ≤12 months before screening or, in the absence of a biopsy with fibrosis, by the presence of two or more HRCT findings, including cystic abnormalities on ≥2 scans, with the most recent performed ≤12 months before screening. The safety and tolerability profile of nintedanib therapy was favourable in children with established fibrosis aged ≥6 years included in InPedILD.97 98 Exploratory analyses of efficacy showed encouraging activity. Although the difference was not significant, patients in the nintedanib group had a mean increase in forced vital capacity of 0.3%±1.3% predicted at 24 weeks compared with a decline of 0.9%±1.8% in the placebo group (nominal p=0.60).97 Bayesian analysis indicated a median difference of 1.63% (95% credible interval –0.69 to 3.40).98
Future directionsClinical and translational investigations into condition-specific development of pulmonary fibrosis in infants and children are needed to define the mostly unknown natural disease course as well as the disease mechanisms in which injury, repair and fibrosis occur in the context of normal lung growth. Transfer of advances already achieved from the intense study of idiopathic pulmonary fibrosis in adults into childhood may speed up the introduction of successful treatments in younger patients.
The definition of fibrosis detailed here and used in InPedILD needs to be further substantiated and may provide a model for future placebo-controlled randomised trials that assess whether the development or progression of pulmonary fibrosis can be prevented. For drug trials in adults that are supported by pharmaceutical companies, investigational plans should be extended to the paediatric age groups, if possible. Similarly, investigator-initiated studies in adults should be re-evaluated for the inclusion of paediatric centres with appropriate expertise.
An exploitable advantage of many of the chILD conditions is knowledge about their molecular basis and disease pathways, which may be targeted by personalised therapy. For example, high-content screening of US Food and Drug Administration-approved small molecules identified ciclosporin A as a potent corrector of some ABCA3 mutation variants.99 Repurposing of approved drugs may help to facilitate treatment options for some conditions including ABCA3 deficiency. Gene therapy is a promising option to treat lung diseases due to monogenetic causes like SP-B-, SP-C and ABCA3 deficiency.100 Use of induced pluripotent stem cell (iPSC) technology (eg, iPSC systems carrying the ILD-associated SFTPC variant) can also be helpful for disease modelling and assessment of novel therapeutics.101
ConclusionsIn the last decade, there have been developments in our understanding of the genetic component of chILD and the progression of certain disorders into adulthood. With the discovery of novel entities, there is a need for improved knowledge of chILD among pulmonologists to optimise the transition of care from paediatric to adult facilities. Updated evidence-based guidelines are needed that incorporate recommendations for the diagnosis and management of immune-mediated disorders, as well as chILD in older children approaching adulthood. Translational and clinical research into the mechanisms of fibrosing pathways in well-defined chILD is critical to develop targeted strategies. Monitoring disease progression and developing a therapeutic strategy to prevent pulmonary fibrosis or its progression are imperative for the future treatment of ILD in children and adolescents, and when they move into adult care.
Ethics statementsPatient consent for publicationAcknowledgmentsThis review was funded by Boehringer Ingelheim International. The authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment related to the development of the manuscript. Writing, editorial support and formatting assistance were provided by Hannah Cook, PhD and John Carron, PhD of Nucleus Global, which was contracted and funded by Boehringer Ingelheim. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
留言 (0)