
記住我

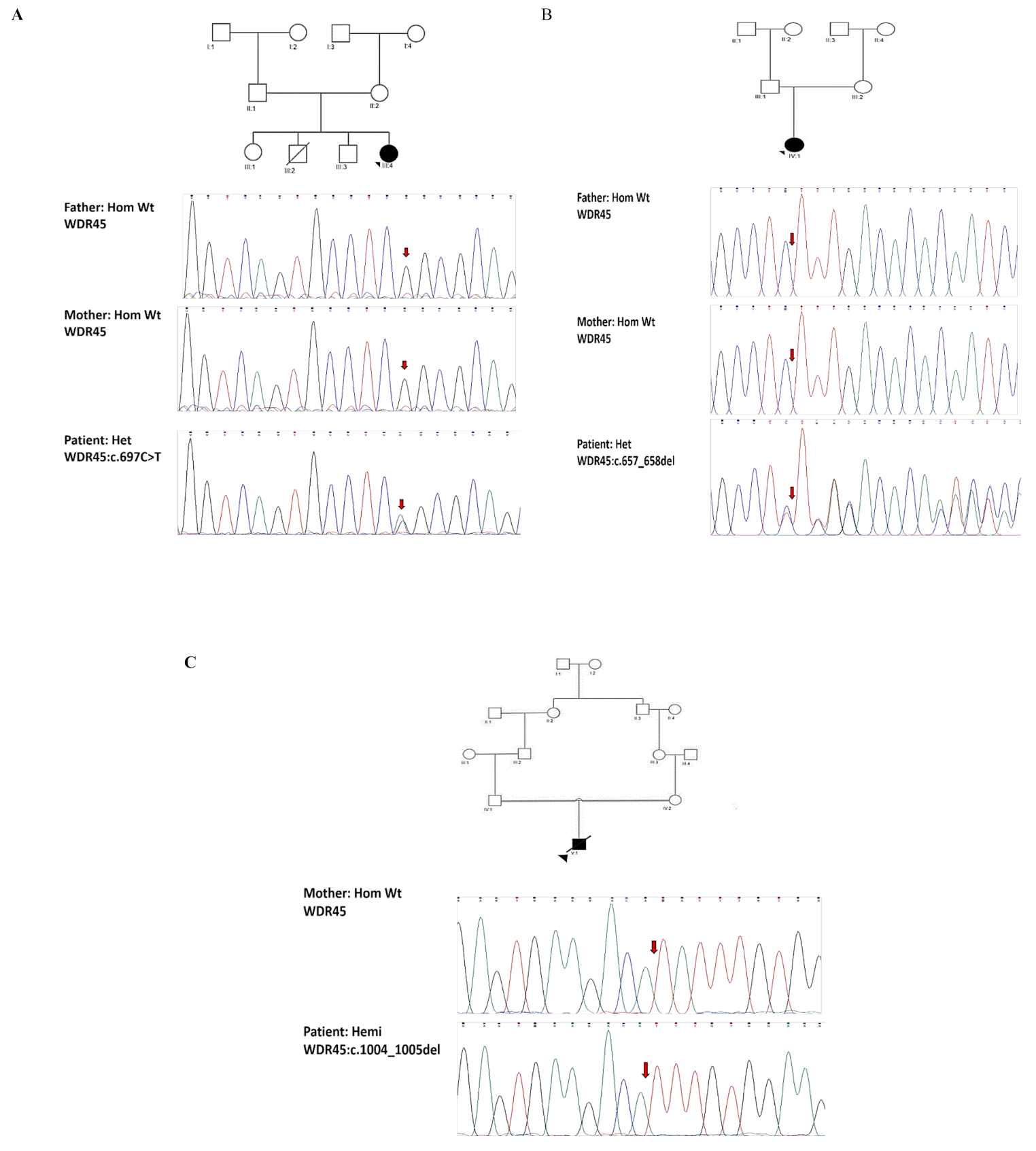

A family suspected of having Leigh syndrome was examined in this study; the main clinical features are shown in Table S1.
First, we examined the proband, an 8-year-old male, born via cesarean section at 36 weeks; he is the second child from the second pregnancy. His birth weight was 2.6 kg. He did not experience perinatal asphyxia but presented with jaundice and received phototherapy. The patient had a normal birth history but developed a high muscle tone at 4 months of age. Compared to his peers, he gradually developed significant motor, language, and cognitive delays as he aged. He could control his head at 3 months, roll over at 4 months, sit independently at 8 months, crawl at 10 months, and stand-alone at around 2 years; he started speaking at 2. At the age of 7, he could walk a few steps independently, speak short phrases, and express basic needs. Also, at the age of 7, he fell while walking, which resulted in a deep chin wound healing slower than usual. Approximately 10 days after the injury, persistent extension and pain occurred in the left limb. After three months of rehabilitation therapy with some improvement, muscle tone significantly increased following cessation of rehabilitation therapy, prompting hospital admission for further evaluation.
Physical examination (at 7 years) showed the patient is in a passive position with involuntary head movements. Horizontal nystagmus was observed in both eyes. The left limb was flexed in a back-extended position, with the left thumb adducted. Muscle tone was high; the patient was uncooperative during muscle strength examination, showing poor muscle coordination in all limbs. Bilateral knee reflexes and Achilles reflexes were slightly hyperactive, and the Babinski sign was negative on both sides.
Cranial magnetic resonance imaging (MRI) findings of the proband revealed multiple symmetric abnormal signals within the cranial cavity. A hyperintense lesion was observed in the right medulla (Fig. 1). Magnetic resonance spectroscopy (MRS) indicated abnormal dual-peak lactate (Fig. 2). Video electroencephalography (EEG) showed slow background activity. During sleep, bilateral frontal 20–25 Hz low-amplitude β waves were detected. One cluster of sleep-related seizures was observed, accompanied by slight tremors in the lower limbs. Concurrent EEG displayed predominant 1 Hz high-amplitude δ slow waves with superimposed low-amplitude 30–35 Hz high-frequency fast waves in the right central, parietal, and midline regions (Cz). Short bursts lasted for 1 s (Fig. 3).
Fig. 1
The proband’s cranial MRI examination. The first diagnosed patient showed multiple symmetric abnormal signals within the intracranial space, with hyperintense lesions on the right side of the medulla. (A) FLAIR: hypointense lesion on the right side of the medulla. (B) T2: hyperintense lesion on the right side of the medulla. (C) FLAIR. (D) T2. (E) FLAIR. (F) T2:hyperintense lesions in the basal ganglia. (G) T1: hypointense lesions on the right side of the medulla. (H) T1: hyperintense lesions in the basal ganglia
Fig. 2
Abnormal MR spectroscopy showing lactic acid double peaks
Fig. 3
Initial patient sleep EEG monitoring. During sleep, seizures occurred, characterized by slight tremors in the lower limbs. Simultaneous EEG recording showed predominant 1 Hz high-amplitude δ slow waves, superimposed with low-amplitude 30–35 Hz low-frequency and high-frequency fast waves in the right central, parietal, and midline areas (Cz) for a brief burst of 1-second
The proband received a cocktail therapy comprising vitamin B, coenzyme Q10, levodopa, and L-carnitine. Significant improvement in muscle tone was observed compared to before treatment. The proband was then discharged from the hospital. The last follow-up was conducted at the age of 8. After discharge from the hospital, the child stopped taking drugs due to vomiting after taking the medication (the medication was taken for about 1 month).
At present, the child is still undergoing rehabilitation treatment, and he has reduced muscle tension. He still has involuntary movements of the head, limbs, and torso and uses his right hand to eat, followed by intentional actions such as biting food, chewing food, other rhythmic movements, and quiet state light (video 1). No seizures were reported. Blood lactic acid was 5.98 mmol/L, blood pyruvate was 0.121 mmol/L, and blood lactic acid/pyruvate was 49.421. His parents are healthy and non-consanguineous (Fig. 4).
Fig. 4
The proband’s older sister is the first child of the first pregnancy, delivered via cesarean section at 36 weeks with a birth weight of 2.8 kg. She had a normal birth history. She exhibited growth and developmental delays around 6 months of age. She could independently sit at around 1 year, having low muscle tone. She was evaluated at 1 year of age at our hospital. Cranial MRI (bilateral medial peduncle lesion, Fig. 5) and genetic metabolic screening (tandem mass spectrometry) in blood and urine were performed, showing no abnormalities. Her cognitive development was stagnant after 7 years of age. Now, the patient is 11 years old. Because of low muscle tone, she cannot walk and stand independently, but she can sit. Obvious involuntary head movements were observed in a sitting position, and paroxysmal horizontal nystagmus and isotropia were also detected, which were very obvious when the patient was nervous (video 2). Blood lactic acid was 5.48 mmol/L, blood pyruvate was 0.115 mmol/L, and blood lactic acid/pyruvate was 47.652.
Fig. 5
The cranial MRI of the proband’s sister. Brain stem lesion. (A, C, E, F) T1: brain stem lesion; (B, D) T2: brain stem lesion
The proband’s younger brother is the third child of the second pregnancy, delivered via cesarean section at 36 weeks with a birth weight of 2.8 kg. He did not experience perinatal asphyxia but had jaundice and received phototherapy. He also experienced developmental delays from a young age; he started walking and speaking at the age of 2.5, although he currently has unclear speech and can express basic needs. He can walk unaided but often struggles. He walks with a wide gate, drags his left lower limb, has bilateral postural incoordination involuntary limb tremors (video 3), and has an intellectual level better than the older children. Blood lactic acid was 4.22 mmol/L, blood pyruvate was 0.089 mmol/L, and blood lactic acid/pyruvate was 47.416.
Whole exome sequencingPeripheral blood samples (2 ml) were collected from the proband, his parents, and brothers, using EDTA as an anticoagulant. The samples were sent to Beijing Maikino Medical Laboratory for genetic testing. Genomic DNA was extracted from white blood cells isolated from peripheral blood using the QIAamp DNA extraction kit (Qiagen, Shanghai, China). The whole genome DNA was fragmented to 150–200 bp by enzyme digestion, and biotin capture probes were employed (Makino, Beijing, China). The gene’s exome and flanking 20 bp regions were enriched to construct the target genome library. Double-ended sequencing was performed using the DNBSEQ-T7 sequencer (BGI, Shenzhen, China), with reads of about 150 bp. Data quality control ensured an average sequencing depth of > 100X, and coverage required that the sequencing depth of 20X was > 95%. The FASTQ file was aligned to the Human Reference Genome (hg19) using BWA software. Sentieon software parameters were utilized to detect SNP and Indel variations. ANNOVAR was employed for SNP/Indel variation annotation. Hazard prediction was performed by association with 1000 genomes, ESP6500, dbSNP, EXAC, HGMD, and REVEL, as well as MutationTaster, SIFT, PolyPhen-2, SPIDEX, and dbscSNV. The pathogenicity of the variants was assessed according to the American Society for Medical Genetics and Genomics (ACMG) guidelines. Association analysis was carried out along the family line to screen for common mutations among the three children, emerging or complex heterozygous variants.
Compound heterozygous mutations were identified in the Ndufa3 gene in proband’s samples (transcript NM_004542), with mutation sites c.10 + 1G > T and c.66_68delCTT (p.22_23delSFinsS). Both parents were heterozygous carriers of these mutations. Siblings were also found with compound heterozygous mutations (Fig. 6).
Fig. 6
First-Generation Sequencing Profiles. (A) Sequencing profile of the proband’s mutation site c.10 + 1G > T. (B) Sequencing profile of the proband’s mutation site c.66_68delCTT. (C) Sequencing profile of the proband’s father’s mutation site c.10 + 1G > T. (D) Sequencing profile of the proband’s father’s mutation site c.66_68delCTT. (E) Sequencing profile of the proband’s mother’s mutation site c.10 + 1G > T. (F) Sequencing profile of the proband’s mother’s mutation site c.66_68delCTT. (G) Sequencing profile of the proband’s sister’s mutation site c.10 + 1G > T. (H) Sequencing profile of the proband’s sister’s mutation site c.66_68delCTT. (I) Sequencing profile of the proband’s brother’s mutation site c.10 + 1G > T. (J) Sequencing profile of the proband’s brother’s mutation site c.66_68delCTT
Minigene Experiment (c.10 + 1G > T)To validate the potential splicing effect caused by the c.10 + 1G > T mutation, an in vitro minigene splicing assay was conducted (Figure S1). Wild-type (WT) and mutant-type (MT) Ndufa3 plasmids were constructed, containing exon1, exon2, and intron1. The WT construct was amplified from normal human genomic DNA using the following primers: Ndufa3 - F5’AAGCTTGGTACCGAGCTCGGATCCGCTGTCGCCGCCGCGGAGACAAAGATGG3’; Ndufa3 - R5’TTAAACGGGCCCTCTAGACTCGAGCGAGGCCCCCGACGACGAAGGACACGAC3’. The amplified product was cloned into the pMini-CopGFP vector (Beijing Hitrobio Biotechnology Co., Ltd., Beijing, China) at the BamHI/XhoI restriction sites using the ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China). WT was validated by Sanger sequencing. MT was obtained by site-directed mutagenesis of WT using the following primers: Ndufa3 - MT-F: 5’ GGCTGCGATTAAGTGCAGGTGCCGGTGGCGCA3’ and Ndufa3 - MT-R: 5’ TGCACTTAATCGCAGCCATCTTTGTCTCCGCG3’. MT was validated by Sanger sequencing. WT and MT plasmids were transfected into HEK293T cells. After 48 h, total RNA was extracted from the cells using TRIzol reagent (Cowin Biotech Co., Jiangsu, China). Reverse transcription polymerase chain reaction (RT-PCR) was performed using primers 5’GGCTAACTAGAGAACCCACTGCTTA3’ and 5’CCCCCGACGACGAAGGACC3’. Gel electrophoresis and Sanger sequencing were used to analyze PCR fragments and determine gene isoforms. Finally, the nucleotide sequence was translated into a protein sequence using the Expasy-translate tool to analyze the impact of the mutation on the translation process.
Investigators concluded the following: ① the wild-type plasmid transcribed mRNA sequence aligns with expectations, containing complete exon 1 and exon 2 (Fig. 7A); ② the mutant-type plasmid transcribes one mRNA product: retention of a 10-bp sequence in the 1st intron, denoted as NM_004542:c.10 + 1_10 + 10insTTAAGTGCAG (Fig. 7B). Without undergoing nonsense-mediated decay (NMD), a frameshift mutation results in a truncated protein (Fig. 7C), represented as p.Arg4IlefsTer59. Overall analysis suggested that the NM_004542:c.10 + 1G > T mutation causes the retention of a 10-bp sequence in the 1st intron (Fig. 7D).
Fig. 7
Minigene experimental results. (A) Wild-type plasmid transcription mRNA sequence matches the expected pattern, including complete exon1 and exon2. (B) Mutant plasmid transcribes a single mRNA product: intron 1 retention with a 10 bp sequence. (C) In the absence of nonsense-mediated decay (NMD), a frameshift results in the formation of a truncated protein. (D) NM_004542: The c.10 + 1G > T mutation leads to intron 1 retention with a 10 bp sequence
Conservation analysis and Gene ExpressionThe transcriptional sites of the Ndufa3 gene NM_004542, specifically c.10 + 1G > T and c.66_68delCTT, exhibit substantial conservation across various species (Figure S2). The gene expression profile of Ndufa3 reveals significant activity during pivotal developmental stages, spanning embryonic, fetal, infant, and adult phases (Figure S3, source: https://gdap.org.cn/). Ndufa3 manifests predominant expression in particular tissues (Figure S4, source: https://gdap.org.cn/), with noteworthy presence in bone marrow, muscles, pituitary gland, prostate, salivary glands, skin, and blood. Additionally, expression is discernible in the brain, highlighting its importance in neural tissues.
留言 (0)