
記住我

The following mouse lines were used in the present study: LepR-Cre (B6.129-Leprtm3(cre)Mgmj/J, the Jackson Laboratory, IMSR cat. no. JAX:032457); PACAP-EGFP (Tg(Adcyap1-EGFP)FB22Gsat/Mmucd, MGI, cat. no. 4846839)75; Rosa26Lox-stop-LoxHTB (the Salk Institute for Biological Studies)76; Vglut2-Cre (Slc17a6tm2(cre)Lowl/J, the Jackson Laboratory, IMSR, cat. no. JAX:016963); Vgat-cre (Slc32a1tm2(cre)Lowl/J, the Jackson Laboratory, IMSR, cat. no. JAX:016962); TrpV1-cre (B6.129-Trpv1tm1(cre)Bbm/J, the Jackson Laboratory, IMSR, cat. no. JAX:017769); Rosa_DTA (Gt(ROSA)26Sortm1(DTA)Jpmb/J, the Jackson Laboratory, IMSR, cat. no. JAX:006331); FosTRAP2 (Fos2A-iCreER/+(FosTRAP2), the Jackson Laboratory, IMSR, cat. no. JAX:030323); and NaV1.3-floxed (B6.129S6-Scn3atm1.1Jwo/H; EMMA strain, cat. no. EM:02214). Heterozygous mice were used for experiments with the exception of the NaV1.3-floxed line where homozygous mice were used to create a conditional knock-out (cKO).
All animal experiments were in accordance with the local ethics committee and governing body (Regierungspräsidium Karlsruhe, Germany) and were approved under protocol nos. G-111/14, G-168/15, G-169/18, G-223/18 and G-181/21. Mice were housed at RT (23 ± 1 °C, unless specified otherwise) in an air-conditioned lab space or animal vivarium with a standard 12-h light:dark cycle and free access to food and water. All genetically modified mice in the present study were on the C57BL/6N background. All studies employed a mixture of male and female mice.
Acclimation protocolMice were divided according to their acclimation status. Acclimated animals were attained by continuous exposure to 36 ± 1 °C for 24 h, 4 d or ≥4 weeks, at a humidity level of 45 ± 5%. Full heat acclimation in rodents is reached after around 4 weeks of habituating the animals to warm temperatures19. We therefore generally—and if not stated otherwise—used mice acclimated for 4–5 weeks, which in the present study we denote as ‘≥4 weeks’. Mice held at RT served as a control (non-acclimated) group. For heat acclimation, mice were placed in a climate chamber (Binder, cat. no. KB720) with free access to food and water. All mice were kept at the standard 12-h light:dark cycle. Mice aged between 7 and 14 weeks were used for heat acclimation. Mice were randomly assigned to the two groups.
Heat endurance assayPrevious studies on the dynamics of acclimation reported that acclimatory homeostasis is reached after 25–30 d whereas short-term acclimation occurs after 2–3 d of acclimation36. At the end of acclimation period, animals were evaluated in a heat endurance assay. The heat endurance assay took place in a similar climate chamber to that used for acclimation where the ambient temperature was set to 39 °C ± 0.5 °C. Animals (aged 11–16 weeks after full acclimation) were transferred immediately from the acclimation chamber to the 39 °C chamber (always in the morning, between 09:00 and 11:00), where they took part in a heat endurance assay lasting for up to a maximum of 24 h. Similar to the acclimation period, mice had free access to food and water. The body temperature of the mice was constantly monitored for the entire period. A body temperature of 41.5 °C was used as the cut-off criterion37,77. At the end of the heat endurance test, animals were shortly placed back to 36 °C to avoid prolonged hypothermia and monitored until the animals were sacrificed. Mice were tested only once in the heat endurance assay and not multiple times.
In experiments where mice where supplemented with leptin during heat acclimation (and before heat endurance assay), animals were administered leptin (Peprotech, cat. no. 450-31, diluted in phosphate-buffered saline (PBS)) at an i.p. dose of 1.25 mg kg−1 twice daily78.
General immunohistochemistry proceduresAnimals were deeply anesthetized with isoflurane and transcardially perfused with PBS (3.85 g of NaOH and 16.83 g of NaH2PO4 in 1 l of distilled water) followed by a 4% paraformaldehyde (PFA) solution. Brains were dissected out and left overnight (O/N) in 4% PFA at 4 °C. Over the next 2 d, brains were immersed into PBS/sucrose solutions (24 h in 10% sucrose followed by 30% sucrose, until the brains sank to the bottom of the container tube). Brains were sectioned with a cryo-microtome at 30-μm thickness and sections (free floating) were kept in cryoprotectant solution (250 ml of glycerol and 250 ml of ethylene glycol made up to 1 l with PBS) at 4 °C until stained.
For antibody staining (ʽAntibodiesʼ), sections were washed once in PBS and left overnight at 4 °C in 0.2% Triton X-100 (PBX0.2). On the following day, sections were blocked with 5% goat serum in PBS containing 0.1% Triton X-100 (PBX0.1) for 2 h at RT. Sections were then incubated with primary antibodies, diluted in 1% goat serum in PBX0.1 for 3 d at 4 °C. On the fifth day, sections were washed extensively with PBX0.1 and then incubated with secondary antibodies and DAPI for 4 h at RT. Finally, tissue was washed extensively with PBSX0.1 and once with PBS, after which sections were mounted using Immu-Mount (Thermo Fisher Scientific, cat. no. 9990402) on to glass slides.
Confocal images were taken at the Nikon imaging center of Heidelberg University, with the Nikon A1R confocal microscope under Nikon Plan Apo λ ×10 magnification, numerical aperture (NA) 0.45 (working distance 4 mm, field of view 1.27 × 1.27 mm2) objective. Cell counting of cells expressing markers of interest was performed with NIS-Elements software (Nikon Instruments, Inc.) using an automatic cell-counting method. The same thresholding of the fluorescence signal was used for each of the color channels in all the quantified images. Images presented were processed with ImageJ.
AntibodiesThe following antibodies were used: chicken anti-GFP (1:1,000, Novus Biotechne, cat. no. NB100-1614); rabbit anti-c-Fos (1:1,000, Synaptic Systems, cat. no. 226 003); rabbit anti-mCherry (1:1,000, Abcam, cat. no. ab167453R); rabbit anti-SCN3A (1:700, Abcam, cat. no. ab65164); goat anti-chicken Alexa Fluor-488 (1:750, Thermo Fisher Scientific, cat. no. A-11039); goat anti-rabbit Alexa Fluor-555 (1:750, Thermo Fisher Scientific, cat. no. A-21430); and DAPI (1:10,000, Sigma-Aldrich, cat. no. 10236276001).
TRAPping of WRNs using FosTrap2 miceTRAPping of WRNsHeterozygous FosTRAP2;HTB mice (resulting from crossing FosTRAP2 mice with the Rosa26Lox-stop-LoxHTB reporter line) were habituated in their home cages in a climate chamber (Binder, cat. no. KB720) at 23 °C and injected with saline solution on 5 d consecutively to reduce stress responses. On the day of the experiment, the climate chamber was warmed to 36 °C; 2 h into warmth exposure, z-4-hydroxytamoxifen (4-OHT) (see below) was delivered by i.p. injection at a dose of 50 mg kg−1. Mice were kept at 36 °C for another 2 or 6 h to reach a total of 4-h and 8-h TRAPping duration, respectively. Control FosTRAP2;HTB mice kept at RT (and not warmed to 36 °C) were treated in the same way (5 d consecutively of saline injections before 4-OHT injection). After the corresponding warmth exposure, both groups of animals were left at thermoneutrality (31 °C) for 48 h to prevent secondary trapping of cold-responsive cells and expecting the 4-OHT to be completely metabolized. For electrophysiology, mice were subsequently either kept at RT or acclimated at 36 °C.
Drug preparation4-OH (Sigma-Aldrich, cat. no. H7904) was prepared for i.p. delivery essentially as described previously79 with some modifications: 4-OHT was dissolved at 20 mg ml−1 in ethanol by vigorous shaking at RT for 5 min + 1 min of sonication in a bath sonicator and was then aliquoted in 50-μl (1-mg) aliquots and stored at −80 °C for up to several weeks. Before use, 4-OHT was redissolved by vigorous shaking at RT for 5 min + 1 min of sonication in a bath sonicator; subsequently, 200 μl of a 1:4 mixture of castor oil:sunflower seed oil (Sigma-Aldrich, cat. nos. 259853 and S5007) was added per 50-μl aliquot containing 1 mg of 4-OHT and the ethanol:oil suspension was vigorously mixed; then, the ethanol was evaporated by vacuum under centrifugation (without heating). The final 5 mg ml−1 of 4-OHT solution was always used on the day of preparation. All injections were delivered intraperitoneally.
For immunohistochemistry, animals following warmth exposure were left at 31 °C until the next day. After this, all three groups (4-h TRAPped, 7-h TRAPped and control groups) were transferred to their home cages for the next 2.5 weeks. After this period, all mice were placed in a climate chamber for 24 h at 23 °C to get accustomed once more to the chamber’s environment. On the next day, the temperature in the climate chamber was adjusted to reach 36 °C to perform the classic warming challenge for hours. After the 4-h exposure to warmth, animals were sacrificed using isoflurane and transcardially perfused. POA-containing brain sections were cut at 30-μm thickness as described above. Tissue was stained for GFP and cFos to quantify the overlap of the TRAP-positive neurons (HTB/GFP positive), with neurons expressing endogenous cFos after the 36 °C warming stimulus.
Expression of cFOS in VMPOLepR neurons after exposure to 36 °C ambient temperatureTo elucidate the role of VMPOLepR neurons in thermoregulatory responses, we investigated whether LepR+ neurons are activated by acute warmth exposure. To do this, LepR-Cre mice crossed to the Rosa26Lox-stop-LoxHTB reporter line80 (here referred to as LepR-Cre;HTB mice) were accustomed to the climate chamber for 24 h. On the second day control animals were taken out, anesthetized with isoflurane and transcardially perfused with PBS, followed by 4% PFA.
The temperature of the climate chamber was switched to 36 °C and the experimental animals were kept at this temperature for 4 h, immediately followed by anesthesia and perfusion. Brains were dissected out and left O/N in PFA at 4 °C. Brains were immersed in sucrose solutions and sliced as described above. α-GFP and α-cFos primary antibodies were applied to amplify HTB/GFP reporter and label endogenous cFos proteins.
NaV1.3 channel stainingC57BL/6 and NaV1.3flox/flox mice were injected with AAV-Cre-GFP. After 4 weeks, to allow AAV expression and protein turnover, animals were transcardially perfused with PFA and brain tissue was processed for immunohistochemistry as described above. Then, 30-μm free-floating brain sections containing POA and cortex were stained with primary antibodies against NaV1.3 and GFP.
Constructs for Scn3a knock-downShRNA constructs for Scn3a were developed according to the method described in ref. 81, with the murine Scn3a canonical complementary DNA sequence as the template. The AAV2-based CAG::FLEX-rev-hrGFP:mir30(Scn9a) vector, used previously by Branco et al.62, was used as a backbone after the excision of the shRNA sequence-targeting NaV1.7 using EcoRI and XhoI restrictases (New England Biolabs). Using the miR_Scan tool (https://www.ncbi.nlm.nih.gov/staff/ogurtsov/projects/mi30), we selected three sequences, binding to the 5ʹ-region (encoding the extracellular loop between segments 5 and 6 of domain I of the channel: sense strand sequence GAAGGACTATATCGCAGATGA), a central region (encoding the intracellular loop connecting domains II and III: sense strand sequence GTGGAGAAATACGTAATTGAT) and the 3ʹ-region (encoding segment 2 of domain IV: sense strand sequence GTCCCGAATCAACCTGGTATTT), to construct shRNAs against. Sense strands and guide strands, separated by the loop sequence TACATCTGTGGCTTCACTA, and supplemented with restriction site overhangs, were synthesized as oligonucleotides and, together with complementary oligonucleotides, aligned and cloned into the recipient vector. Such AAV vectors, where the shRNA sequences were placed between a FLEX switch sequence together with a GFP reporter gene, were packaged into AAV1/2 particles by the Viral Vector Facility, University of Zurich (Switzerland).
As a negative control for these shRNA Scn3a constructs, we produced an AAV containing a scrambled sequence (ACTGTAGTCGTCGACTTACCAT) that was subcloned into the same vector backbone as functional shRNAs.
AAV brain injectionsAll surgical procedures were performed under aseptic conditions and deep anesthesia. Adult mice (7–18 weeks) were anesthetized using an i.p injection of anesthesia mix (medetomidine 0.5 mg kg−1, midazolam 5 mg kg−1 and fentanyl 0.05 mg kg−1). Mice were placed on a stereotaxic apparatus (Model 1900, Kopf) and kept warm using a heating pad at 33.5 °C. The fur of the head was removed, the skin disinfected (Braunol, Braun) and the cornea moisture preserved during surgery by the application of eye ointment (Bepanthen, Bayer). Craniotomies of approximately 0.5-mm diameter were drilled on the skull with a hand drill (Osada Electric, cat. no. OS40). A pulled-glass capillary with a 20- to 40-µm tip diameter was lowered into the brain and specific recombinant AAV (rAAV) carrying the functional construct or a fluorescent protein was injected using a manual air pressure system.
The following AAVs and titers were used:
single-stranded (ss)AAV-DJ/2-hSyn1-chI-dlox-hChR2(H134R)_mCherry(rev)-dlox-WPRE-hGHp(A) (Zurich Vector Core, 5.3 × 10E12 vg ml−1)
ssAAV-DJ/2-hSyn1-chI-dlox-mCherry(rev)-dlox-WPRE-hGHp(A) (Zurich Vector Core, 7.2 × 10E12 vg ml−1)
ssAAV-5/2-hSyn1-chI-dlox-EGFP_2A_FLAG_TeTxLC(rev)-dlox-WPRE-SV40p(A) (Zurich Vector Core, 7.7 × 10E12 vg ml−1)
ssAAV-1/2-hEF1α-dlox-hM4D(Gi)_mCherry(rev)-dlox-WPRE-hGHp(A) (Zurich Vector Core, 4.5 × 10E12 vg ml−1)
ssAAV-1/2-hSyn1-chI-dFRT-EGFP_2A_FLAG_TeTxLC(rev)-dFRT-WPRE-hGHp(A) (Zurich Vector Core, 5.0 × 10E12 vg ml−1)
ssAAV-1/2-hSyn1-dlox-EGFP(rev)-dlox-WPRE-hGHp(A) (Zurich Vector Core, 6.7 10E12 vg ml−1)
ssAAV2/9-CAG::FLEX-rev-hrGFP:mir30(Scn9a) (a gift from S. Sternson, 1.5–1.7 10E13 GC per ml)
ssAAV2/9-CAG::FLEX-rev-hrGFP:mir30(Scn9a-scrambled) (a gift from S. Sternson, 1.5–1.7 10E13 GC per ml)
ssAAV-1/2-hEF1α-dlox-hM3D(Gq)_mCherry(rev)-dlox-WPRE-hGHp(A) (Zurich Vector Core, 4.0 × 10E12 vg ml−1)
ssAAV-retro/2-hSyn1-chI-dlox-mCherry_2A_FLPo(rev)-dlox-WPRE-SV40p(A) (Zurich Vector Core, 6.3 × 10E12 vg ml−1)
ssAAV-retro/2-hSyn1-chI-dlox-EGFP_2A_FLPo(rev)-dlox-WPRE-SV40p(A) (Zurich Vector Core, 9.9 × 10E12 vg ml−1)
ssAAV-1/2-hSyn1-dFRT-hM4D(Gi)_mCherry(rev)-dFRT-WPRE-hGHp(A) (Zurich Vector Core, 8.4 × 10E12 vg ml−1)
ssAAV-1/2-shortCAG-dlox-miR(NaV1.3-v1)(rev)-hrGFP(rev)-dlox-WPRE-SV40op(A) (Zurich Vector Core, 1.0 × 10E13 vg ml−1)
ssAAV-1/2-shortCAG-dlox-miR(NaV1.3-v2)(rev)-hrGFP(rev)-dlox-WPRE-SV40op(A) (Zurich Vector Core, 8.9 × 10E12 vg ml−1)
ssAAV-1/2-shortCAG-dlox-miR(NaV1.3-v3)(rev)-hrGFP(rev)-dlox-WPRE-SV40op(A) (Zurich Vector Core, 7.8 × 10E12 vg ml−1)
ssAAV-1/2- shortCAG-dlox-miR(NaV1.3-scrambled)(rev)-hrGFP(rev)-dlox-WPRE-SV40op(A) (Dirk Grimm laboratory, Heidelberg University, 1.9 × 10E12 vg ml−1)
ssAAV-8/2-CAG-EGFP_Cre-WPRE-SV40p(A) (Zurich Vector Core, 2.1 × 10E12 vg ml−1)
ssAAV-1/2-hSyn1-chI-iCre-WPRE-SV40p(A) (Zurich Vector Core, 5.2 × 10E12 vg ml−1)
ssAAV.DJ/2.hEF1α.dlox.GCaMP6f(rev).WPRE.bGHp(A) (Zurich Vector Core, 4.8 × 10E12 vg ml−1).
Skin was sutured with sterile, absorbable, needled sutures (Marlin, cat. no. 17241041, catgut) and mice were injected subcutaneously with carprofen at 5 mg kg−1 (Rimady, Zoetis). Finally, anesthesia was antagonized using a subcutaneous injection of atipamezole 2.5 mg kg−1, flumazenil 0.5 mg kg−1 and naloxone 1.2 mg kg−1 and mice were transferred to their home cages. For postoperative care, a second dose of carprofen was injected after 24 h and mice cages were kept on a veterinary heating pad at 37 °C for 12 h and monitored closely. A minimum of 3 weeks of viral expression was allowed before any experiments were conducted.
Telemetry transmitter implantationAll animals (with the exclusion of those used for electrophysiological recordings) were implanted with a telemetry transmitter (Data Sciences International, cat. no. TA11TA-F10) to monitor body temperature during the acclimation procedure and behavioral testing. Animals were injected intraperitoneally with an anesthesia mix as described above, and the fur of the abdomen was removed, the skin disinfected with Braunol (Braun, cat. no. 3864065) and the cornea protected with Bepanthen ointment (Bayer). A sterile telemetric transmitter was implanted in the abdominal cavity. Thereafter, muscle and skin layers were separately sutured with absorbable surgical threads. After the surgery, the anesthesia was antagonized and animals were monitored as described above; recovery for at least 1 week was allowed before any further procedures were undertaken.
Tail, interscapular BAT and core body temperature measurementIn ChR2-encoding, AAV-injected mice (and respective control animals), tail temperatures and BAT temperatures were measured using an infrared thermal camera (VarioCAMhr, InfraTec). Snapshot images were taken every 5 min using IRBIS 3 software (InfraTec). The average temperature was calculated in the middle of the tail (segment length of 1 cm) and at the center of the interscapular region, which was shaved 3–5 d before measurement. Core body temperature was sampled every 5 min via receiver plates (DSI, cat. no. RSC-1) placed underneath the cages. Telemetry data were registered using Ponemah (DSI). All measurements were conducted during the light phase.
Optogenetic stimulation of LepR cellsStereotactic surgeries were performed in adult LepRCre neurons. Animals were injected bilaterally with 250 nl of AAV encoding the Cre-dependent ChR2 or mCherry (control group) (ssAAV-DJ/2-hSyn1-chI-dlox-hChR2(H134R)_mCherry(rev)-dlox-WPRE-hGHp(A) or ssAAV-DJ/2-hSyn1-chI-dlox-mCherry(rev)-dlox-WPRE-hGHp(A)) at coordinates targeting VMPO neurons: bregma: mediolateral (ML): ±0.400 mm, anteroposterior (AP): 0.800 mm, dorsoventral (DV): −4.850 mm (VMPO). A 200-μm diameter fiberoptic probe (ThorLabs, cat. no. FT200UMT) was lowered to target the preoptic LepR cell population (coordinates: bregma: ML: 0.400 mm, AP: 0.800 mm, DV: −4.700 mm (VMPO)). The probe was anchored to the skull with dental acrylic. After the surgery, the anesthesia was antagonized and mice were transferred to their home cages. Postoperative care and telemetry implantation were performed as described above. At least 4 weeks were allowed for recovery and full expression of ChR2 before the start of optogenetic stimulation.
To activate ChR2-expressing LepR neurons, a fiberoptic probe was attached through an FC/PC adapter to a 473-nm blue light-emitting diode (LED; Optogenetics-LED-Blue, Prizmatix). All experiments were conducted unilaterally and the fiberoptic cable was connected at least 2 h before the experiments to allow for habituation. For the optogenetic probing, mice received light pulses of 4–6 mW power and 10 ms, delivered at 20-Hz stimulation frequency using a Prizmatix Pulser software and pulse train generator. In each optogenetic probing experiment, the light stimulation period was of 1 min followed by an interstimulation interval of 3 min.
TeTxLC and Gi-DREADD silencing of LepR cellsFor these experiments, stereotactic surgeries were performed in adult LepRCre mice as described in previous sections; 250 nl of rAAV encoding the Cre-dependent tetanus toxin light chain (TeTxLC) (ssAAV-5/2-hSyn1-chI-dlox-EGFP_2A_FLAG_TeTxLC(rev)-dlox-WPRE-SV40p(A)) or the inhibitory Gi-DREADDs (ssAAV-1/2-hEF1α-dlox-hM4D(Gi)_mCherry(rev)-dlox-WPRE-hGHp(A)) was injected bilaterally into the VMPO. AAVs encoding a Cre-dependent mCherry/EGFP were used as controls. After brain injection, the anesthesia was antagonized and mice were transferred to their home cages. Postoperative care was performed as described above. After telemetry implantation, heat (or RT) acclimation and heat endurance assay were performed as detailed in previous sections.
Acute chemogenetic silencing of LepR cells was performed by i.p. injection of CNO (or saline) 0.3 mg kg−1 (Enzo, diluted in saline) 10 min before transferring the animals to the heat endurance assay. Body temperature was constantly monitored as mentioned above. To validate CNO effects on the firing frequency of acclimated LepR cells, a group of chemogenetically silenced animals were used for in vitro electrophysiological recordings. The slice preparation and electrophysiological recording procedures are described below.
Gq-DREADD and ChR2 stimulation and long-term activation (optogenetic and chemogenetic conditioning) of LepR cellsFor these experiments, stereotactic surgeries were performed in adult LepR-Cre mice as described in previous sections; 250 nl of AAV encoding for Cre-dependent excitatory Gq-DREADDs (ssAAV-1/2-hEF1α-dlox-hM3D(Gq)_mCherry(rev)-dlox-WPRE-hGHp(A)) or Cre-dependent ChR2 (ssAAV-DJ/2-hSyn1-chI-dlox-hChR2(H134R)mCherry(rev)-dlox-WPRE-hGHp) was injected bilaterally into the VMPO.
To mimic acclimation by optogenetic activation of VMPOLepR cells, ChR2-expressing mice received light pulses via the fiberoptic probe of 10-ms duration or pulse (4–6 mW), triggered at 1-Hz frequency. The stimulation was protracted continuously for the duration of the heat challenge (maximum 9 h) or was started 1 d or 3 d before heat endurance. The degree of hypothermia produced by this continuous optogenetic stimulation was tested in a different cohort of mice at RT.
For chemogenetic activation of VMPOLepR cells to mimic acclimation, animals were injected daily with CNO (i.p. 0.3 mg kg−1, Enzo, diluted in saline) for 1, 5 or 10 d consecutively. The CNO effect on their body temperature was monitored constantly. At the end of the injection period, and 24 h from the last injection, animals were tested in the heat endurance assay while the temperature was recorded telemetrically as described above. A group of long-term, chemogenetically stimulated animals were used for electrophysiological recordings.
Repeated administration of CNO to control mice (in the absence of Gq-DREADD) over a period of 10 d also had a small but discernible effect, in particular on the kinetics of Tcore at the initial phase of the heat endurance assay, possibly reflecting the activity of CNO metabolites, such as clozapine, known to modulate several neuronal receptor systems82,83. Nevertheless, this effect was considerably smaller than that observed in mice carrying the chemogenetic activator Gq-DREADD. Of note, all mice carrying Gq-DREADD and chemogenetically conditioned for 10 d reached the cut-off time in the heat endurance assay, suggesting that their gained heat tolerance—and different to that of any of the other experimental groups—was underestimated in this assay (Extended Data Fig. 9c,d).
Silencing of LPBN neuronsVglut2-Cre mice were injected with 250 nl of retroAAV encoding the Cre-dependent FlpO recombinase (ssAAV-retro/2-hSyn1-chI-dlox-mCherry_2A_FLPo(rev)-dlox-WPRE-SV40p(A) or ssAAV-retro/2-hSyn1-chI-dlox-EGFP_2A_FLPo(rev)-dlox-WPRE-SV40p(A)) bilaterally into the VMPO. After 3 weeks, the same animals received 250 nl of a bilateral injection of AAV encoding the FlpO-dependent TeTxLC or the inhibitory Gi-DREADD (ssAAV-1/2-hSyn1-chI-dFRT-EGFP_2A_FLAG_TeTxLC(rev)-dFRT-WPRE-hGHp(A) or ssAAV-1/2-hSyn1-chI-dFRT-EGFP_2A_FLAG_ hM4D(Gi)(rev)-dFRT-WPRE-hGHp(A)) at bregma: ML: ±1.25 mm, AP: −4.900 mm, DV: −2.7 mm (LPBN). After telemetry implantation, heat acclimation was performed as detailed earlier.
Acute chemogenetic silencing of LPBN presynaptic partner cells was performed by i.p. injection of CNO (or saline) 0.3 mg kg−1 (Enzo, diluted in saline) at the end of the acclimation protocol and 10 min before transferring the animals to the heat endurance assay. Cre-negative animals were subjected to the same injection procedure and served as controls. Body temperature was constantly monitored for all animals during the acclimation period and/or the heat endurance assay.
NaV1.7 or NaV1.3 knock-down in VMPOLepR cellsAnimals were anesthetized as described above and the shRNA virus against Scn9a or NaV1.7 (rAAV2/9-CAG::FLEX-rev-hrGFP:mir30(Scn9a)) or scrambled control (rAAV2/9-CAG::FLEX-rev-hrGFP:mir30 (Scn9a-scrambled))62 or against Scn3a or NaV1.3 (ssAAV-1/2-shortCAG-dlox-miR(NaV1.3-v1/v2/v3)(rev)-hrGFP(rev)-dlox or scrambled control ssAAV-1/2-shortCAG-dlox-miR(NaV1.3-scrambled)(rev)-hrGFP(rev)-dlox) was injected into the POA to target LepR+ neurons (250 nl, bilaterally). The three NaV1.3-targeting shRNA AAVs (v1, v2 and v3) were mixed at 1:1:1 proportions before injections. After recovery and acclimation, animals were used for in vitro electrophysiological recordings.
NaV1.3 cKOTo create NaV1.3 cKO, NaV1.3-floxed mice were brought to homozygosity (NaV1.3fl/fl) and injected with Cre-encoding AAV (AAV8 CAG EGFP-Cre) into VMPO. Wild-type mice (NaV1.3+/+) were injected with Cre-encoding AAV to serve as controls. At least 3 weeks of virus expression or protein turnover was allowed before subjecting the cKO mice and controls to heat acclimation. A mix of Cre-AAV-injected wild-type (WT) littermates and WT C57BL/6 mice was used as a control group.
Histology of AAV-injected mouse brainsMice were anesthetized, transcardially perfused with PFA and decapitated. The entire heads were left in 4% PFA for at least 1 d at 4 °C. Subsequently, the brains were removed from the skull and transferred to PBS containing sucrose. Coronal sections of 30 µm were cut at the microtome and stored at −20 °C in cryoprotectant solution. Subsequently, brain sections were stained for GFP or mCherry as described previously.
LepR cell dissociation (for RNA-seq)Adult mice LepR-Cre;HTB, acclimated and non-acclimated (10–12 weeks of age), were anesthetized with isoflurane and decapitated. The brain was immediately removed and submerged in ice-cold artificial cerebrospinal fluid (aCSF). Three brains were sectioned at the same time on a Vibratome (Leica, cat. no. VT1200S) in a slicing chamber containing ice-cold aCSF: NaH2PO4 (1.2 mM), KCl (1.2 mM), Hepes (20 mM), glucose (25 mM), NaHCO3 (30 mM), N-methyl-d-glucamine (NMDG; 93 mM), Na ascorbate (5 mM), Na pyruvate (3 mM), N-acetylcysteine (12 mM), CaCl2 (0.5 mM) and MgSO4·7H2O (10 mM), constantly bubbled with carbogen. Brain slices of 250-μm thickness, containing the rostral POA, parts of the OVLT and MnPO, were transferred to a Petri dish containing aCSF. We implemented the neuron isolation protocol described in ref. 20. The regions of interest (ROIs)were micro-dissected under a dissecting microscope and transferred to a small Petri dish containing 3 ml of papain mix consisting of Hibernate mix (Hibernate-A medium (Invitrogen, cat. no. A1247501), 1× Glutamax (Gibco, cat. no. 35050-038), 0.8 mM kynurenic acid (Sigma-Aldrich, cat. no. K3375-5G), 0.05 mM AP-V (HelloBio, cat. no. HB0225), 0.01 mM Rock inhibitor Y-27632 (HelloBio, cat. no. HB2297), 1 mM B27 (Invitrogen, cat. no. 17504001), 5% trehalose (Sigma-Aldrich, cat. no. T9531-10G)) and 8 U ml−1 of papain (Sigma-Aldrich, cat. no. P4762), 100 U ml−1 of DNAse I (Worthington über Cell-Systems, cat. no. LK003172), 0.005 U ml−1 of chondroitinase ABC (Sigma-Aldrich, cat. no. C3667-5UN), 0.07% hyaluronidase (Sigma-Aldrich, cat. no. H2126) and 0.001 mM NaOH. The tissue was cut into smaller pieces and transferred together with the papain mix into a 2-ml tube at 37 °C to incubate while shaking (700 r.p.m.) for 2 h. After incubation the papain solution was pipetted out of the tube and exchanged with Hibernate mix containing 0.1 mg ml−1 of ovalbumin and centrifuged for 1 min at 300g. Supernatant was removed and the Hibernate mix was added to the tissue pieces, which were further dissociated into single cells by gentle trituration through Pasteur pipettes with fire-polished tip openings of 600-, 300- and 150-μm diameter. Cell suspension was centrifuged at RT and 300g for 10 min and the supernatant was removed and exchanged with 500 μl of Hibernate-A medium. Resuspended cell material was passed through a 20-µm filter. Cell suspension was stained with propidium iodide (PI; BD Pharmingen, cat. no. 5166211E) to exclude the dead cells before the FACS analysis. FACS sorting was performed on a BD FACS Aria II using the purity sorting mode. FACS populations were chosen to select cells with low PI and high GFP fluorescence.
Cells were FACS sorted into bulks of GFP+ and GFP− directly into the RLT buffer (QIAGEN RNeasy Micro Kit, cat. no. 74004), according to the arbitrary levels of GFP fluorescence, immediately frozen on dry ice and stored at −80 °C. Samples were processed for a maximum of 1 month from the isolation by using the column purification method according to the manufacturer’s instructions and samples were stored at −80 °C until further processing.
cDNA library preparation (for RNA-seq)RNA integrity and the concentration of each sample were assessed by Agilent Bioanalyzer Nano 6000 chip (Agilent Technologies) and QUBIT (Invitrogen, cat. no. QUBIT2) measurement. We used the Smart seq2 protocol84 for the cDNA library preparation (all processing performed at Gene Core EMBL, Heidelberg). Then, 200 pg of each RNA bulk sample was processed for the reverse transcription (Superscript IV) followed by 18 cycles of PCR amplification, library tagmentation (Tn5 transposase produced in house, PEP Core EMBL, Heidelberg), sample barcoding and a final 12 cycles of PCR enrichment. All samples were sequenced on Illumina NextSeq 500 High sequencer, single end with 75-bp long reads (Gene Core EMBL).
The RNA sequencing (RNA-seq) results are deposited at Array Express (https://www.ebi.ac.uk/biostudies/arrayexpress) and can be found under the following accession no.: E-MTAB-14029
POA slice preparation for electrophysiologyFor in vitro electrophysiology, 8- to 15-week-old mice were deeply anesthetized using a ketamine/xylazine mixture (ketamine: 220 mg kg−1 (Ketavet, Zoetis) and xylazine 16 mg kg−1 (Rompun, Bayer)), decapitated and their brains transferred to ice-cold (4 °C) oxygenated (95% O2, 5% CO2) slicing aCSF (in mM): NaCl, 85; KCl, 2.5; glucose, 10; sucrose, 75; NaH2PO4, 1.25; NaHCO3, 25; MgCl2, 3; CaCl2, 0.1; myoinositol, 3; sodium pyruvate, 2; and ascorbic acid, 0.4. Coronal (250-μm thick) POA slices were prepared with a Vibratome and then incubated at 32 °C in a bath containing oxygenated holding aCSF (in mM): NaCl, 109; KCl, 4; glucose, 35; NaH2PO4, 1.25; NaHCO3, 25; MgCl2, 1.3; and CaCl2, 1.5. After a recovery period of 30 min, individual slices were transferred to the recording chamber where they were continuously superfused with oxygenated recording aCSF (for recipes, see below) at ~2 ml min−1.
In some experiments, brain slices were prepared using carbogen-bubbled NMDG–Hepes solution (at 4 °C) containing (in mM): NMDG, 93; KCl, 2.5; NaH2PO4, 1.2; l(+)-ascorbic acid, 5; thiourea, 2; sodium pyruvate, 3; MgSO4·7H2O, 10; CaCl2·2H2O, 0.5; Hepes, 20; NaHCO3, 30; glucose, 25; and N-acetyl-l-cysteine, 10 (pH 7.37–7.38, 295–305 mosmol kg−1). After slicing, POA coronal slices were incubated for 15 min in the same NMDG–HEPES solution at 32 °C and subsequently transferred to a chamber containing holding aCSF composed of (in mM): NaCl, 118; KCl, 2.5; NaHCO3, 24; NaH2PO4, 1.2; sodium pyruvate, 2.4; l(+)-ascorbic acid, 4; N-acetyl-l-cysteine, 2; Hepes, 5; MgSO4, 1; CaCl2, 2; and glucose, 7 (pH 7.3–7.5, 295–305 mosmol kg−1).
Cells in acute POA slices were visualized using a SliceScope upright microscope (Scientifica) equipped with a ×40 water immersion objective (Olympus, cat. no. U-TV1X-2). Images were acquired by a digital CCD camera (Hamamatsu Photonics K.K., ORCA-R2, cat. no. C10600-10B) using MicroManager 1.4 software (Vale’s lab, University of California San Francisco (UCSF)). Electrophysiological recordings were acquired using a MultiClamp 700B amplifier (Molecular Devices), together with an Axon Digidata 1550B digitizer (Molecular Devices) and Clampex 11.0.3 software (Molecular Devices). All signals were sampled at 20 kHz and low pass filtered at 10 kHz. Borosilicate glass micropipettes used (outer diameter 1.5 mm, inner diameter 0.86 mm; Sutter Instrument, cat. no. BF150-86-7.5) were pulled on a micropipette puller (Sutter Instrument, cat. no. P-97). Intracellular solution was passed through a 0.22-µm filter before filling the electrode pipette. The open pipette resistance was between 4 MΩ and 8 MΩ.
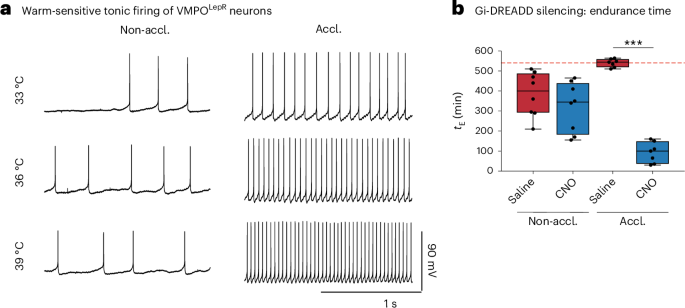
Electrophysiological measurement of warmth sensitivity of VMPO neuronsIn acute slice experiments where neuronal action potentials were recorded at varying temperatures, a bridge in a form of glass capillary filled with agar dissolved in 3 M KCl was placed between the bath chamber and the ground electrode to isolate the reference electrode from the temperature changes applied to the chamber85. Equipment for bath temperature control consisted of temperature-controlled microscope stage (Luigs & Neumann, cat. no. TC07), an in-line heater (Warner, cat. no. CL-100) and a liquid cooling system (Warner, cat. no. LCS-1).
Neuronal action potentials were recorded with aCSF containing (in mM)—NaCl, 125; KCl, 6.25; glucose, 15; NaH2PO4, 1.25; NaHCO3, 25; MgCl2, 1.3; and CaCl2, 2.4 (called ‘high-K+ aCSF’) as previously described85—and with an internal solution containing (in mM): K gluconate, 138; KCl, 2; NaCl, 5; Hepes, 10; (ethylenebis(oxonitrilo))tetra-acetate (EGTA), 10 (or equimolar amount of BAPTA); CaCl2, 1; and Mg-ATP, 1.
AP frequencies were analyzed in traces where the bath temperature was 33, 36 or 39 °C; a deviation of a maximum ±0.5 °C was tolerated. Neurons were classified as warm sensitive (WSN) when their temperature coefficient reached 0.75 Hz per °C and as cold-sensitive (CSN) when their temperature coefficient was lower than −0.6 Hz per °C, thresholds traditionally used to define central temperature-sensitive neurons86. Temperature-insensitive neurons had their temperature coefficient between ≥−0.6 Hz per °C and <0.75 Hz per °C and neurons were classified as silent when not a single spontaneous AP could be detected. Cells unable to produce AP even when stimulated with current injection were excluded from analysis. Probing VMPO neuronal populations for temperature sensitivity was done in the presence of synaptic blockers (gabazine 5 µM, CNQX 10 µM and AP-V 50 µM) added to the bath solution. In experiments where the effect of cholinergic transmission was tested, 10 μM tubocurarine and 10 μM scopolamine were included in the perfusion fluid.
In some experiments, APs were measured without varying bath temperature (at 33 °C) and with a ‘low-K+ aCSF’, containing (in mM): NaCl, 125; KCl, 2.5; NaHCO3, 24; NaH2PO4, 1.2; Hepes, 5; MgSO4, 1; CaCl2, 2; and glucose, 8. The solution used and temperature of recordings are indicated in the figure legends showing spontaneous AP firing data.
Recordings of ionic currentsThe RMP was measured in the current-clamp mode using extracellular solution containing (in mM): NaCl, 150 (or equimolar amount of NMDG); KCl, 3.5; Hepes, 10; glucose, 20; CaCl2, 1.2; and MgCl2, 2 (as per ref. 87). TTX (0.5 μM) was added to the aCSF and pipette solution contained (in mM): K gluconate, 120; Hepes, 40; MgCl2, 5; Na2ATP, 2; and Na-GTP, 0.3.
To record voltage-ramp responses in voltage-clamp mode to approximate passive membrane permeability to potassium, we used a low-sodium and 0 mM nominal calcium solution that contained (in mM): NMDG, 125; NaHCO3, 24; KCl, 2.5; NAH2PO4, 1.2; Hepes, 5; glucose, 8; and MgSO4, 1. The pipette solution contained (in mM)—cesium methanesulfonate, 120; Hepes, 40; MgCl2, 5; Na-ATP, 2; Na-GTP, 0.3; QX-314, 5; tetraethylammonium chloride (TEAC), 5; and 4-aminopyridine (4-AP), 1—to block voltage-gated potassium and sodium channels. ‘Leak’ potassium channels are largely unaffected by intracellular cesium88.
Voltage ramps as well as voltage-gated calcium currents were recorded using the same cesium methanesulfonate-based pipette solution and external solution composed of (in mM): NaCl, 125; NaHCO3, 24; KCl, 2.5; NaH2PO4, 1.2; Hepes, 5; glucose, 8; MgSO4, 1; and CaCl2, 2. To record voltage-gated calcium currents, aCSF additionally contained 0.5 μM TTX, 1 mM TEAC and 100 μM 4-AP.
To measure voltage-gated sodium currents in whole-cell and nucleated patch configurations, we used solutions as described in ref. 89. In the present study, external solution contained (in mM): NaCl, 124; KCl, 3; glucose, 30; NaH2PO4, 0.5; NaHCO3, 25; MgSO4, 1; and CaCl2, 1.5; with the addition of TEAC (5 mM) and CdCl2 (50 μM). The pipette solution contained (in mM): Cs gluconate, 100; NaCl, 4; TEAC, 10; 4-AP, 5; EGTA, 10; CaCl2, 1; Hepes, 10; Mg-ATP, 4; Na-GTP, 0.3; and Na phosphocreatine, 4.
Spontaneous synaptic currents were recorded with ‘low-K+ aCSF’ and cesium methanesulfonate-based pipette solution. The spontaneous excitatory postsynaptic currents were recorded while holding the neurons at −65 mV in gap-free mode; The spontaneous inhibitory postsynaptic currents were recorded at the potential of 0 mV (reversal potential for α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid currents).
In vitro validation of DREADD receptor function was performed in current-clamp mode using low-K+ aCSF, a K gluconate-based intracellular solution as described above and with the addition of 5 µM CNO.
For AP frequency quantification, the first 3 min of recordings were omitted in voltage-clamp recordings and at least 1 min was allowed after break-in before any recording was performed. All ionic current recordings were conducted at 36 °C (±0.5 °C) to mimic more closely the physiological neuronal conditions. Basic cell membrane properties such as capacitance and input resistance were calculated based on a membrane test protocol (a brief step of −10 mV from a holding potential of −65 mV). Series resistance (Rs) was typically 10–25 MΩ across experiments. In voltage-clamp recordings, whole-cell capacitance compensation was applied and Rs values were compensated 50–60%; the compensation was readjusted before each protocol. The voltage protocols applied are shown in the insets to the Extended Data Figs. In current-clamp experiments, pipette capacitance neutralization and bridge balance were used. In experiments where voltage-gated sodium currents were measured, a liquid junction potential (LJP) of 8 mV was corrected online; in other experiments, the LJP was corrected offline. In voltage-clamp experiments, cells with a membrane resistance changed by >50% or Rs values changed by >20% between the start and end of the recording were excluded from analysis. All electrophysiology data were acquired with pClamp 10 and pClamp 11 software (Molecular Devices). An in-house software was developed for the automated analysis of the AP waveforms. Cells were chosen for patch-clamp recordings on a random basis, provided that they were within the specified brain region and had a healthy cell membrane.
Quantitative PCRThe animals were sedated with isoflurane and sacrificed via cervical dislocation 3 weeks after injection of shRNA AAVs to the POA. The whole brain was prepared and stored in cold Dulbecco’s PBS (Gibco). The brain was cut with the help of a mouse brain matrix and the whole POA was extracted and transferred to an Eppendorf tube, which was subsequently filled with TRIzol reagent. RNA was extracted using the TRIzol (Ambion, cat. no. 15596026) and ROTI phenol/chloroform/isoamyl alcohol (Carl Roth, cat. no. A156) protocol. The POA tissue was transferred from TRIzol solution to a glass mortar and manually disrupted with a pestle. Subsequently, disrupted tissue was suspended in ROTI phenol/chloroform/isoamyl alcohol. The samples were centrifuged at 208 r.c.f. (relative centrifugal force) in a tabletop centrifuge at 4 °C for 10 min. The resulting aqueous phase was transferred to a spin column for purification (Zymo Research, cat. no. R1013) and the eluted RNA was stored at −80 °C until further analysis.
Total RNA, 600 ng, was used for first-strand cDNA generation with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific, cat. no. 10368252) using oligo(dT) primers according to the manufacturer’s instructions. The resulting cDNA was diluted to a concentration of 600 ng µl−1. The cDNA was analyzed by qPCR using the following primers specific for the Scn3a transcript. Ube2l3 and Tubb3 served as housekeeping genes. Primer sequences are listed here:
Gene
Primer sequence
Scn3a
F: GTGGACCTGGGCAATGTCT
R: CACGATGGTCTTTAAACCTGGAA
Ube2l3
F: CAGCAGCACCAGATCCAAGA
R: GGTTGTCAGGAACAATAAGCCC
Tubb3
F: TGAGGCCTCCTCTCACAAGT
R: GTCGGGCCTGAATAGGTCTC
The qPCR amplification reactions (15 µl) contained 7.5 µl of FastStart Essential DNA Green Master Mix (Roche, cat. no. 06402712001), 5 µl of RNase-free water (QIAGEN), 1 µl of cDNA and 1.5 µl of forward (F) and reverse (R) primer. Reactions were run on a Roche LightCycler 96 System (Roche Diagnostics). Controls without reverse transcription were included to control for traces of genomic DNA. No template controls were included to check for contamination and nonspecific amplification. The resulting CT values were exported as text files and imported into Microsoft Excel for further analysis. The acquired data were analyzed by an approach described in refs. 90,91. Data are expressed as relative gene expression ratios. All samples were measured in triplicates.
Microendoscopy of VMPOLepR neurons in awake behaving mice (Miniscope experiments)Experiments were performed in 8-week-old, male LepR-Cre+/− mice. Each mouse underwent two sequential stereotaxic surgeries, one for injections of an AAV vector expressing GCaMP6f (Zurich Virus Core) and a second performed 7 d later to implant a gradient refractive index (GRIN) lens attached to a baseplate.
For AAV injections, mice were deeply anesthetized with 2% isoflurane at a flow rate of 0.5 l min−1 and placed in a stereotactic frame (Kopf Instruments). Body temperature was maintained at 37 °C with a heating pad (Hot-1, Alascience, Scientific Instruments). Ophthalmic ointment (Bepanthen) was applied to the eyes to prevent drying. On deep anesthesia, mice underwent bilateral craniotomies at two AP locations, using a high-speed, rotary, micro-drill (Stereotaxic Drill, Kopf Instruments). The following stereotaxic coordinates were used: 0.2 mm AP ± 0.4 mm ML and 0.5 mm AP ± 0.4 mm ML. Then, a glass pipette filled with GCaMP6f delivered the virus into the VMPO (5-mm DV). In each injection site, 200 nl of a 1:3 virus dilution in saline solution was injected, using a NanoJet microinjector (World Precision Instruments) at a rate of 20 nl min−1. After each injection, the pipette was left at the injection site for 10 min to avoid backflow and then slowly withdrawn. The skin was then sutured with nylon suture thread (Dafilon, Braun).
At 7 d, a GRIN lens + baseplate was implanted. For this, mice were again anesthetized as previously described and a 0.6 × 7 mm2 GRIN lens with an integrated baseplate for the nVista, miniaturized, head-mounted microscope (Miniscope, Inscopix) was slowly inserted into the brain (60 µm min−1) on one of the hemispheres previously injected. The following stereotaxic coordinates were used: 0.35 mm AP, 0.4 mm ML and 5 mm DV. Then, the baseplate was fixed to the skull using a self-curing adhesive resin (Super-Bond, Sun Medical) and a light-cured composite resin (Gradia-Direct Flo, GC Corp.). The surface of the lens was then covered with a plastic basecap (Inscopix) and the skin was sutured with nylon suture thread.
Then, 6–7 weeks after implantation, the pre-acclimation recordings were performed. To dock the Miniscope to the baseplates, mice were briefly anesthetized with isoflurane (2%). On docking, the Miniscope was locked to the baseplate with a small screw. Mice recovered from anesthesia for at least 30 min, at RT, in a Plexiglass box (25 × 25 cm2) with bedding. Recordings of calcium signals were first performed at RT, at five different focal points. At each focal point, calcium transients were recorded for 2 min, at 20 Hz, with a maximum resolution of 1,280 × 800 pixels2 and an LED power of 1.0–1.1 mW mm−2. After this, mice were moved to the heating chamber that was maintained at close to 36 °C (±2 °C). Recordings at high temperature started 5 min after the mice were transferred into the heating chamber, using the same parameters of low temperature recordings. Once the pre-acclimation recording session finished, mice were temperature acclimated for 30 d, as described earlier. Post-acclimation recordings were performed following the same procedure as for the pre-acclimation recordings described above. In the control experiment, mice were recorded at identical time points and in identical conditions, but maintained at RT between recording sessions (30 d).
After recordings, mice were anesthetized with an i.p. injection of Narcoren, transcardially perfused with 4% PFA in PBS and post-fixed in 4% PFA for 48 h at 4 °C. After this, the implant was removed, the brain dehydrated in 30% sucrose overnight at RT and cut into 30-μm sections using a sliding microtome (Hyrax S50 and KS34, Zeiss). Four series were generated for each mouse’s VMPO and one of these series was mounted and imaged with an EPI fluorescence microscope (×10, Leica, cat. no. DM6000) to assess virus expression and implant location within the MPOA. Raw recordings were first preprocessed using the Inscopix Data Processing Software (IDPS, v.1.6.0.3225). For each experimental session, video-recordings obtained at 22 and 36 °C were merged and processed together. The Timeseries module of the IDPS toolbox was used to merge the two videos. Raw imaging data were cropped to accommodate only the desired ROI and remove the lens boundary artifact. Recordings were then filtered using the spatial filter module that removes the low and high spatial frequencies (measured as the number of oscillations per pixel), thus effectively reducing out-of-focus background fluorescence. We used a trial-and-error method to identify the filter cut-off values and found that the best low cut-off value was 0.005 and the high cut-off value 0.900. The file was then motion corrected using the motion correction module of IDPS. The mean image of the video file was considered as the global reference. The maximum translational value for a pixel was also estimated using the trial-and-error method and it varied between 20 and 40 for recordings from different mice. These files were then exported as .tiff image stacks and were used to extract calcium transients using EZcalcium92 extraction and an analysis toolbox based on the CaImAn pipeline93. For this, we first performed manual ROI detection using the ROI dete
留言 (0)