
記住我

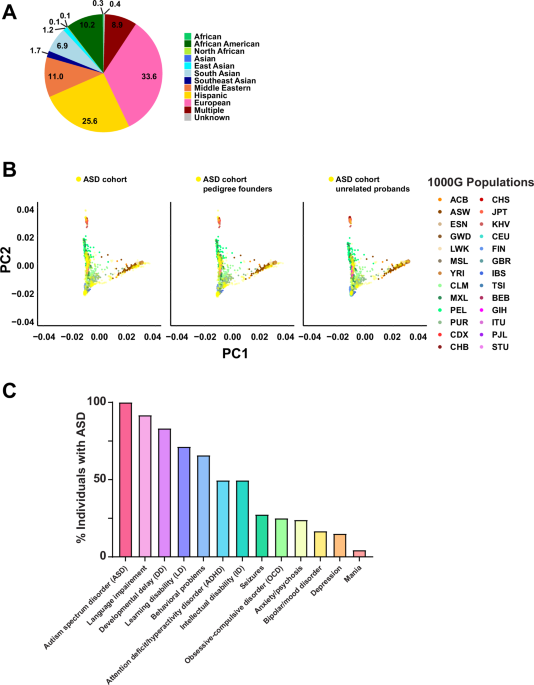
A total of 195 simplex and multiplex families who have at least one child diagnosed with ASD were enrolled in our study (Supplementary Data 1). The enrolled families represent diverse ancestral backgrounds, including African American, Asian, Hispanic, Middle Eastern, Native American, and European (Fig. 1A). We used principal component analysis (PCA) to explore the ancestry of the families in the cohort (Fig. 1B). Our cohort clustered across the different subpopulations of the 1000 Genomes project (1000G)12. Given that our cohort does not comprise a specific population, this finding is consistent with expectations. The cohort included a total of 222 individuals with ASD and their family members without ASD (165 fathers, 188 mothers, 5 grandmothers, and 174 siblings), and we observed a male-to-female ratio of 2.7:1 (162 males, 60 females) among individuals with ASD. This is slightly lower than the more recent estimates of ~3:113,14 or previous estimates of ~4:113. Parental age, which is a possible risk factor for ASD15, was not significantly different at the time of birth of individuals with ASD compared to offspring with no ASD (Supplementary Fig. 1). A standardized medical questionnaire was collected from each of the 195 participating families and reviewed along with available medical records for the presence of clinical comorbidities commonly associated with ASD and other neurodevelopmental disorders, including attention deficit/hyperactivity disorder (ADHD), language delay or impairment, cognitive impairment including intellectual disability, specific learning disability, aggression or challenging behaviors, mood disorders (i.e., anxiety, depression, obsessive-compulsive disorder (OCD), bipolar disorder), seizures, and sleep problems. There were 222 individuals diagnosed with ASD and 532 participants without ASD. Of those individuals with ASD where complete information for a specific phenotype was available, 91.72% had language impairment, 83.21% had developmental delay, 71.31% had learning disability, 65.81% had behavioral problems, 49.55% had ADHD, 49.54% had intellectual disability, 27.45% had seizures, and 25% had OCD (Fig. 1C). Other medical comorbidities were seen at lower frequencies, including environmental and food allergies, and respiratory, gastrointestinal, and vision problems. Demographics and clinical information for the cohort are provided in Fig. 1, Table 1 and Supplementary Data 1.
Fig. 1: Ancestral diversity and phenotypic spectrum of the ASD cohort.
A Pie chart depicting the ancestral diversity of the ASD cohort. Multiple refers to individuals with multiple ancestries. B Principal component analysis (PCA) of the ASD cohort samples combined with the 1000G populations, using the entire ASD cohort (left), the pedigree founders (middle), or the unrelated probands (right). The ASD cohort is represented in yellow. The 1000G populations are: ACB African Caribbeans in Barbados, ASW Americans of African Ancestry in Southwest USA, ESN Esan in Nigeria, GWD Gambian in Western Divisions in Gambia, LWK Luhya in Webuye, Kenya, MSL Mende in Sierra Leone, YRI Yoruba in Ibadan, Nigeria, CLM Colombians from Medellin, Colombia, MXL Mexican Ancestry from Los Angeles, USA, PEL Peruvians from Lima, Peru, PUR Puerto Ricans from Puerto Rico, CDX Chinese Dai in Xishuangbanna, China, CHB Han Chinese in Beijing, China, CHS Southern Han Chinese, JPT Japanese in Tokyo, Japan, KHV Kinh in Ho Chi Minh City, Vietnam, CEU Utah Residents (CEPH) with Northern and Western European Ancestry, FIN Finnish in Finland, GBR British in England and Scotland, IBS Iberian Population in Spain, TSI Toscani in Italia, BEB Bengali from Bangladesh, GIH Gujarati Indian from Houston, Texas, ITU Indian Telugu from the UK, PJL Punjabi from Lahore, Pakistan, STU Sri Lankan Tamil from the UK. Population abbreviations are also defined in Supplementary Data 11. C The prevalence of neurodevelopmental and neuropsychiatric conditions in the ASD cohort. ASD was diagnosed in all 222 probands (100%). Language impairment was the most commonly reported phenotype (91.72%).
Table 1 Demographics and clinical information for the ASD cohortWhole exome sequencing and variant discovery in the ASD cohortWe performed WES on samples from 754 individuals, including 222 individuals with ASD. The average read depth was 46X, with no differences in depth of sequencing with respect to phenotypic status, sex, or family relationships (Supplementary Fig. 2A–C). On average, 99.29% and 93.9% of bases were covered at a mean read depth of at least 10X and 20X, respectively (Supplementary Fig. 2D). An average of 86,215 total variants were identified per exome, of which an average of 73,132 were single nucleotide variants (SNVs) and 13,083 were insertions or deletions (indels) (Supplementary Data 2). After applying read depth and quality filters, 77,075 variants per exome remained, of which an average of 65,907 were SNVs and 11,168 were indels (Supplementary Data 2). A detailed summary of our WES data processing and variant filtration pipeline is shown in Fig. 2. We filtered for rare variants with a minor allele frequency (MAF) < 1% in all annotated population databases ((1000G)12, Genome Aggregation Database (gnomAD)16,17, the Greater Middle East Variome project (GME)18, and The Exome Aggregation Consortium (ExAC)19), identifying on average 8433 rare variants per exome, of which 7002 were heterozygous and 1431 were homozygous (Supplementary Data 2). We defined potentially damaging variants as the subset of rare exonic or splice site (referred to as coding) variants that are also predicted to be damaging by at least 1 of the 2 algorithms used: SIFT and PolyPhen-2 HumVar. There was no significant difference in the number of potentially damaging variants between sexes for individuals with ASD in the cohort (Supplementary Fig. 3). To assess for an excess of potentially damaging variants in individuals with ASD compared to individuals without ASD, we performed a burden analysis. We found no difference between individuals with or without ASD in the burden of rare variants with total coding, nondisrupting, missense damaging, or loss of function effects (Supplementary Fig. 4). This outcome is expected, given our modest sample size and the fact that ASD comprises individually rare diseases with genetic heterogeneity, caused by rare alleles of substantial impact. Therefore, observing an excess of these variations requires studying much larger cohorts capable of capturing this heterogeneity. We discovered an average of 5959 novel variants per exome that have not been reported in any of the populations in the public databases that we used for annotation (Supplementary Data 2). Furthermore, we found an average of 52 novel variants per individual that were private (71 for parents, 34 for offspring), meaning they have not been reported in any of the annotated populations and they were not present in any other individual in the cohort (Supplementary Data 3). In total, there were 38,834 novel private variants across all individuals in the cohort (Supplementary Data 3). As expected, more private variants were present in parents compared with offspring (Supplementary Fig. 5). We identified an average of 15 (20 for parents, 9 for offspring) private coding variants per exome, of which an average of 6 (8 for parents, 4 for offspring) per exome were nonsynonymous and predicted to be potentially damaging by at least 1 of the 2 algorithms used, SIFT and PolyPhen-2 HumVar (Supplementary Data 3).
Fig. 2: Overview diagram of study analyses.
Whole exome sequencing (WES) was performed on 754 individuals from 195 families, including 222 probands with ASD and their family members without ASD (165 fathers, 188 mothers, 5 grandmothers, and 174 siblings). Single nucleotide variants (SNVs) and small insertions or deletions (indels) were called using DeepVariant. Variant quality filtering was performed as described in the Materials and Methods. Rare de novo or inherited (X-linked, homozygous, and compound heterozygous) variants were annotated to identify potentially pathogenic variants. Risk genes were prioritized by disease annotation, specific expression, and pathway enrichment. MAF minor allele frequency. This figure was created with BioRender.com.
Identification of candidate ASD variantsFor candidate ASD variant discovery, we initially focused on rare nonsynonymous exonic or splice site variants that were either de novo or segregated with ASD in the family under homozygous, compound heterozygous, or X-linked inheritance. We identified an average of 4 de novo variants (2 coding) per offspring with ASD (Supplementary Data 4). In addition, we identified an average of 155 inherited homozygous variants (38 coding) and 10 compound heterozygous variants in 3 genes per offspring with ASD (Supplementary Data 4). We also identified an average of 16 recessive X-linked variants in male offspring with ASD (8 coding) (Supplementary Data 4). We did not find a significant correlation between the number of de novo variants and maternal or paternal age at birth of an offspring with ASD (Supplementary Fig. 6). In total, we identified 630 genes harboring 1503 rare nonsynonymous exonic or splice site variants that are predicted to be potentially damaging by at least 1 of the 2 algorithms used, SIFT and PolyPhen-2 HumVar (Supplementary Data 5). The shared symptoms among individuals with ASD suggest the existence of a functional convergence downstream of loci that contribute to the condition. To investigate if there is selective expression of at least some of these 630 genes in different brain regions, we conducted specific expression analysis (SEA) using human transcriptomics data from the BrainSpan collection20. We found that genes with variants detected in the individuals with ASD in our cohort were enriched in the thalamus (p = 0.014) (Fig. 3 and Supplementary Data 6), including AR, ATP1A3, SCN1A, and SLC7A3.
Fig. 3: Enrichment of the identified ASD genes in the thalamus.
Bullseye plot of specific expression analysis (SEA) of genes harboring the prioritized variants across brain regions and development. SEA revealed that genes with possibly damaging variants detected in the ASD cohort were enriched during young adulthood in the thalamus. The color bar shows Benjamini–Hochberg corrected p.
Variants in known ASD or neurodevelopmental disease genesTable 2 summarizes the potentially pathogenic variants in 73 known ASD or neurodevelopmental disease genes for each individual with ASD after variant prioritization. Out of these genes, 40 are reported in the Simons Foundation Autism Research Initiative (SFARI) Gene database21, and the rest are OMIM-annotated disease genes associated with relevant phenotypes, including neurodevelopmental disorder, intellectual disability, developmental delay, and epilepsy. These genes were significantly enriched in pathways involving nervous system development, neurogenesis, and neuronal differentiation (Supplementary Data 7). We identified 92 unique variants in 68 individuals with ASD (~1–3 per individual). Twenty-six individuals with ASD had coding variants in 19 syndromic ASD genes: CDKL5 (3 probands), DMD (3 probands), BCORL1 (2 probands), and SETD1B (2 probands). ARID1B, ATP1A3, CHAMP1, CNOT1, FRMPD4, HUWE1, KAT6A, KMT2C, MECP2, PACS2, PHF21A, SCN1A, SLC6A1, SMARCA2, TFE3, and ZMYM3 are other syndromic ASD genes harboring variants in single probands. Twenty-three individuals with ASD had coding variants in 21 nonsyndromic ASD genes having a SFARI Gene21 score of 1 or 2: NEXMIF (2 probands) and NLGN4X (2 probands). AR, ARHGEF10, ASTN2, AUTS2, BIRC6, CACNA1F, DLG4, DYNC1H1, IL1RAPL1, ITPR1, OPHN1, PCDHA5, SKI, SLC7A3, SYN1, TOP2B, WNK3, YEATS2, and ZC3H4 are other ASD genes harboring variants in single probands. Thirty-two probands had other coding variants in 33 neurodevelopmental disease genes, with 2 genes—ADGRV1 and ATP7A—having variants in 2 probands each. ACSL4, ARHGAP31, ARMC9, ATP2B3, ATP6AP2, BCAP31, CCDC22, CHD5, DBR1, DCTN1, DHX37, FGD1, HDAC6, IGBP1, KIF1C, MINPP1, MPDZ, NOTCH1, NRG1, OBSL1, PIGG, PLXNA1, SAMD9L, SCN3A, SLC13A3, SRPX2, TMEM151A, TNRC6A, TRIM71, TRNT1, and ZNF148 are other neurodevelopmental disease genes harboring variants in single probands. Three probands had coding variants in two neurodevelopmental genes each: MC-159-5 (ADGRV1 and KIF1C), MC-161-3 (MPDZ and NRG1), and MC-172-3 (OBSL1 and SAMD9L).
Table 2 Potentially pathogenic variants in known ASD and neurological disease genes identified in individuals with ASD from the cohortVariants in new candidate ASD genesWe identified 158 potentially pathogenic coding variants in 120 candidate ASD genes after variant prioritization (Table 3). Gene ontology analysis revealed that several of the candidate ASD genes are involved in signal transduction and synaptic activity such as DLG3, GABRQ, KALRN, KCTD16, P2RX4, PKP4, SLC8A3, and TENM2 (Supplementary Data 7). Multiple variants were observed in candidate genes: ATG4A, CNGA2, CROCC, FAM47C, FRMPD3, GABRQ, GPRASP1, MAGEC3, MXRA5, OR5H1, PWWP3B, SLITRK4, TRPC5, TSPYL2, and ZNF630. Since we observed more than one potentially pathogenic variant (in known and/or novel genes) in some probands, we also ranked them according to their likelihood of causing the disease in the proband (Supplementary Data 8). In proband MC-017-3, there were two variants found in SCN1A and RBMX2. The SCN1A variant was prioritized over the RBMX2 variant as SCN1A is a known ASD gene, according to the SFARI Gene database21. Similarly, in proband MC-174-3, a variant in HUWE1, a known neurodevelopmental disease gene22,23, was ranked above a variant in another known neurodevelopmental disease gene ATP6AP224,25 based on AlphaMissense scores, and above a variant in the novel gene MTM1.
Table 3 Potentially pathogenic variants in novel candidate ASD genes identified in individuals with ASD from the cohortCopy number variant analysisSince CNVs are known to play an important role in ASD26, we analyzed CNVs in the ASD cohort. We called CNVs in individuals with ASD using individuals from the cohort who did not have ASD as controls, utilizing CNVkit27. In total, we identified 539 CNVs across all individuals with ASD, including 276 deletions and 263 duplications (Supplementary Data 9 and 10). The average size of a CNV was 243 kb, and there were 15 CNVs encompassing regions that did not include any genes. Out of the identified CNVs, 34 overlapped with known ASD CNVs as defined by the SFARI Gene database21, including the 3q29, 17p11.2, and 22q13.3 loci. Of the called CNVs, 23 also overlapped with syndromic CNVs from the DECIPHER database28. Some of these syndromes, such as Potocki-Lupski syndrome29 and Smith-Magenis syndrome30, are associated with neurodevelopmental phenotypes. Although our data demonstrate an overlap between CNVs and specific genomic regions, this does not imply that the CNVs are causal. Further investigation is needed to establish the pathogenicity of these variants.
留言 (0)