
記住我
Neutrophils are essential innate immune system components, crucial for defending against pathogens including bacteria, virus and fungus (1). The homeostasis of neutrophils production and elimination is critical for appropriate immune system function. Neutrophils originate from multipotent hematopoietic stem cells located in the bone marrow or spleen. These stem cells undergo a series of maturation processes and eventually develop into mature neutrophils that are released into the bloodstream (2). Neutrophils production and maturation are regulated by various factors, including cytokines like CXCL8, growth factors like G-CSF, and cellular signals like PI3K/AKT pathway (3). Granulocyte-stimulating factor (G-CSF) is a key cytokine that enhances neutrophil production by promoting hematopoietic stem cell differentiation and proliferation (4). Mature neutrophils released into the bloodstream form the peripheral neutrophil reserve, which migrates to sites of infection or inflammation (5). On detection of invading pathogens or inflammatory signals, circulating neutrophils adhere to vascular endothelial cells and migrate into tissue spaces, guided by chemotactic factors, where they perform essential functions, such as bactericidal activity and phagocytosis (5).
Neutrophils generally have a relatively short lifespan, with half-lives ranging from several hours to a few days (6, 7). Timely elimination of aged, surplus, or non-functional neutrophils through cell death is crucial for maintenance of appropriate immune responses to various conditions (5, 8). Alteration of neutrophil cell death mechanisms can result in various pathological conditions, including acute inflammation, chronic inflammation and disruption of other immunological pathways (9–12). For example, delayed neutrophil death can exacerbate lung injury in patients with sepsis (13), while suppressed cell death within the microenvironment of certain solid tumors accelerates cancer metastasis (14). Therefore, understanding the detailed mechanisms underlying cell death is pivotal to full comprehension of a wide range of diseases.
Apoptosis, a programmed form of cell death commonly observed during inflammation, is the primary cell death mechanism that aids in clearance of unwanted neutrophils to prevent excessive inflammation (8, 15). However, various other types of neutrophil death have recently been detected, including autophagy, pyroptosis, necrosis, necroptosis, and newly-discovered mechanisms, such as ferroptosis (16–19). In addition to these conventional pathways, neutrophils undergo a specialized form of cell death, which is closely tied to their function, referredas NETosis (20, 21). These different cell death types have distinct roles in various physiological and pathological conditions, including inflammatory, non-inflammatory, pathogen-induced, and cell lysis-mediated contexts (22). It is crucial to determine the predominant mode of cell death occurring under specific conditions. Neutrophils that undergo programmed cell death are phagocytosed and cleared by surrounding macrophages, a process known as efferocytosis, which prevents secondary inflammation or autoimmune responses (23). Further, neutrophils undergoing other forms of cell death are cleared through dissolution and absorption, minimizing their potential harmful effects.
1.2 Neutrophilic function and diseaseNeutrophils both function as first-line effectors in the innate immune response, defending against pathogens, such as bacteria, viruses, and fungi, and have roles in regulating various immunological processes, in contexts including cancer, injury, and tissue repair (24). Neutrophils can synthesize and release lysosomal enzymes, generate oxygen radicals, and release cytokines, underpinning their unique and crucial roles in the immune system (24–26). Due to their diverse functions and regulatory abilities, neutrophils have varying roles in the development of different diseases.
Neutrophils primarily function in protection against infections and tissue damage (24). As major components of the innate immune system, neutrophils eliminate pathogens through phagocytosis and by releasing toxic and antibacterial substances. Further, these cells can modulate immune responses by controlling the timing of pro- or anti-inflammatory cytokine release, thereby regulating inflammation intensity, to effectively remove infections without impairing tissue repair (26).
Abnormal neutrophil activity and numbers can have negative effects on disease development (24). Insufficient neutrophil activity increases susceptibility to opportunistic infections and allows infection spread. Conversely, excessive neutrophil infiltration can result in overwhelming inflammation, which delays tissue regeneration and increases the risk of reinfection (27). Further, dysregulated neutrophil activation can lead to overproduction of pro-inflammatory factors or insufficient anti-inflammatory factor generation, contributing to tissue damage in various organs and autoimmune diseases; for example, rheumatoid arthritis and Crohn’s disease (12, 24). Neutrophils are also crucial components of the tumor immunological microenvironment, where their functions and activities undergo specific modulation (28). In some cases, neutrophils aid tumor cell recognition and killing, while in others they promote tumor growth, metastasis, and drug resistance (28, 29). The mechanisms underlying the effects of neutrophils on tumors are complex and varied, and specific tumor cell types can exploit neutrophil immunoregulatory mechanisms to evade immune surveillance and accelerate tumor progression (30).
1.3 Immunometabolism and neutrophil deathCell metabolism, which involves energy provision and biomolecule synthesis, is crucial for most cellular processes. Immunometabolism, referring to integration of metabolism in the immune system, has been a focus of immunology research for some years. Both intercellular and intracellular metabolic signaling strongly influence the activation state and fate decisions of immune cells, including neutrophils. Neutrophils can rapidly transition from quiescent to activated states, which requires dynamic metabolism to meet their energy and biosynthetic demands. Metabolic signaling operates bidirectionally, in that cellular signaling can reprogram metabolism, while metabolites play critical roles in multiple signaling pathways. Specific cellular processes trigger the activation of distinct metabolic pathways, enabling cell adaption, underscoring the critical role of metabolic fluctuations (31). Neutrophils have traditionally been considered glycolysis-dependent cells that primarily derive energy from glucose (32, 33). However, recent studies have highlighted the importance of other metabolic pathways, including mitochondrial, lipid, and glutamine metabolism, in the neutrophil lifespan and their functions (34–37).
Few studies to date have linked neutrophilic metabolism with cell death, and the comprehensive metabolic features associated with specific types of cell death remain unclear. Reactive oxygen species (ROS) have pivotal roles in various forms of cell death (38–40). utilize multiple metabolic pathways to generate ROS, which can be synthesized in the cytoplasm or mitochondria (17, 34, 35, 41). Nevertheless, the precise mechanisms governing ROS production and utilization in specific types of neutrophil cell death remain incompletely elucidated, and warrant further research. Furthermore, the processes of neutrophil death can be influenced by metabolites other than ROS (41, Table 1).
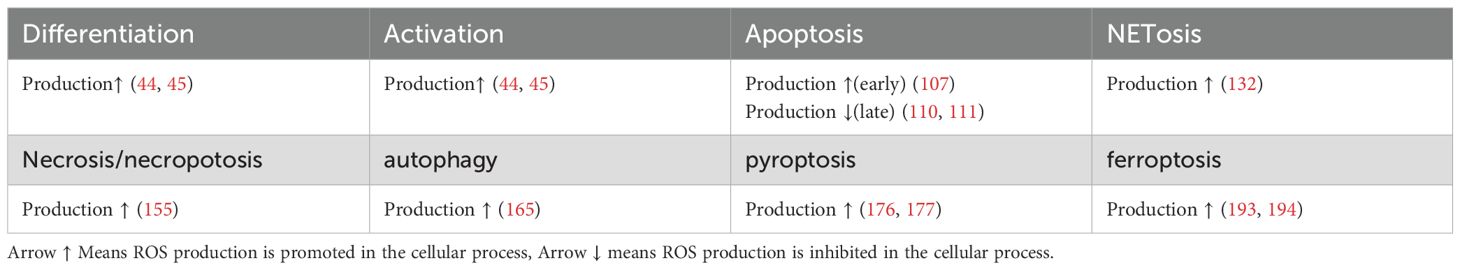
Table 1. ROS production in different processes in neutrophils.
In this review, we provide an overview of the fundamental metabolic profiles of neutrophils in both quiescent and activated phases. Additionally, we summarize the known relationships among neutrophil metabolic reprogramming and various cell death processes. Our aim is to outline a concise map of metabolic transitions throughout the lifespan of neutrophils and offer insights that can inspire future research (Table 2).
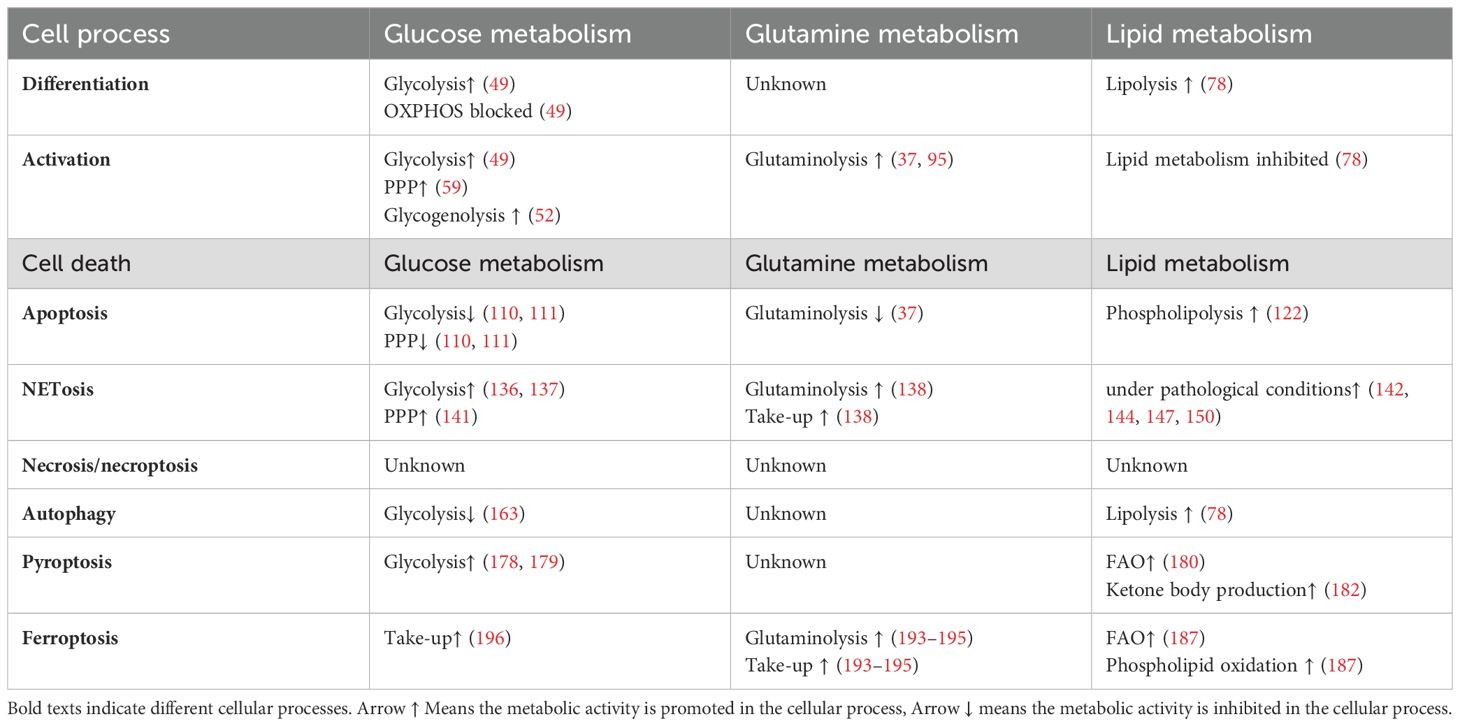
Table 2. Metabolism alteration in different processes in neutrophils.
2 Fundamental metabolism profile of neutrophilsIn this section, we discuss the primary metabolic pathways in neutrophils. Although neutrophils rely on glycolysis to provide most of the energy required for their basic functions (32, 33), different types of metabolism are activated in neutrophils during phagocytosis and other immunological processes (34, 42, 43). Oxygen consumption and ROS production are remarkably promoted as neutrophils transition into an active state, and this phenomenon is referred to as respiratory burst (44, 45). Glycolysis and other neutrophilic metabolic processes are influenced by respiratory burst, despite glycolysis generally being considered an anaerobic pathway (46). Other metabolic alterations also warrant deep investigation, to facilitate comprehensive understanding of neutrophil metabolism.
2.1 Glucose metabolismGlucose is the major substance used to supply energy in neutrophils (34). Neutrophils obtained from the surrounding environment via the glucose transport proteins, GLUT1, GLUT3, and GLUT4, which are expressed on neutrophil cytomembranes (47, 48). When neutrophils are in a quiescent state, GLUT1 serves as the chief glucose transporter; however, on activation, there is a marked increase in GLUT3 and GLUT4 expression, which ensures sufficient glucose supply (48).
As progenitor myeloid cells differentiate into neutrophils, their main energy source shifts from oxidative phosphorylation (OXPHOS), which connects the tricarboxylic acid (TCA) cycle with the electron transport train in aerobic respiration, to glycolysis (49). This transition allows neutrophils to function effectively in inflammatory environments, which are typically low in available oxygen, and is triggered by hypoxia inducible factor-1α (HIF-1α). Hypoxia inhibit the hydroxylation of H1F-1α by prolyl hydroxylase domain family and factor inhibiting HIF (FIH) dioxygenases to permit function of HIF-1α. HIF-1α is a transcriptional factor that promote the expression of glucose transporters like GLUT1 and GLUT3 and glycolytic enzymes like aldolase and phosphoglycerate kinase-1 (50). HIF-1α can also suppress OXPHOS byenhancing the expression of pyruvate dehydrogenase kinase 1and and LDHA to prevent pyruvate from being turned into actyl-CoA that enter TCA cycle. Once glucose enters neutrophils, it is immediately converted into glucose-6-phosphate (G6P), to prevent it leaving the cell. Under aerobic conditions, G6P is transformed into lactate, the ultimate product of glycolysis, rather than remaining as pyruvate for entry into the TCA cycle (33, 51) (Figure 1).
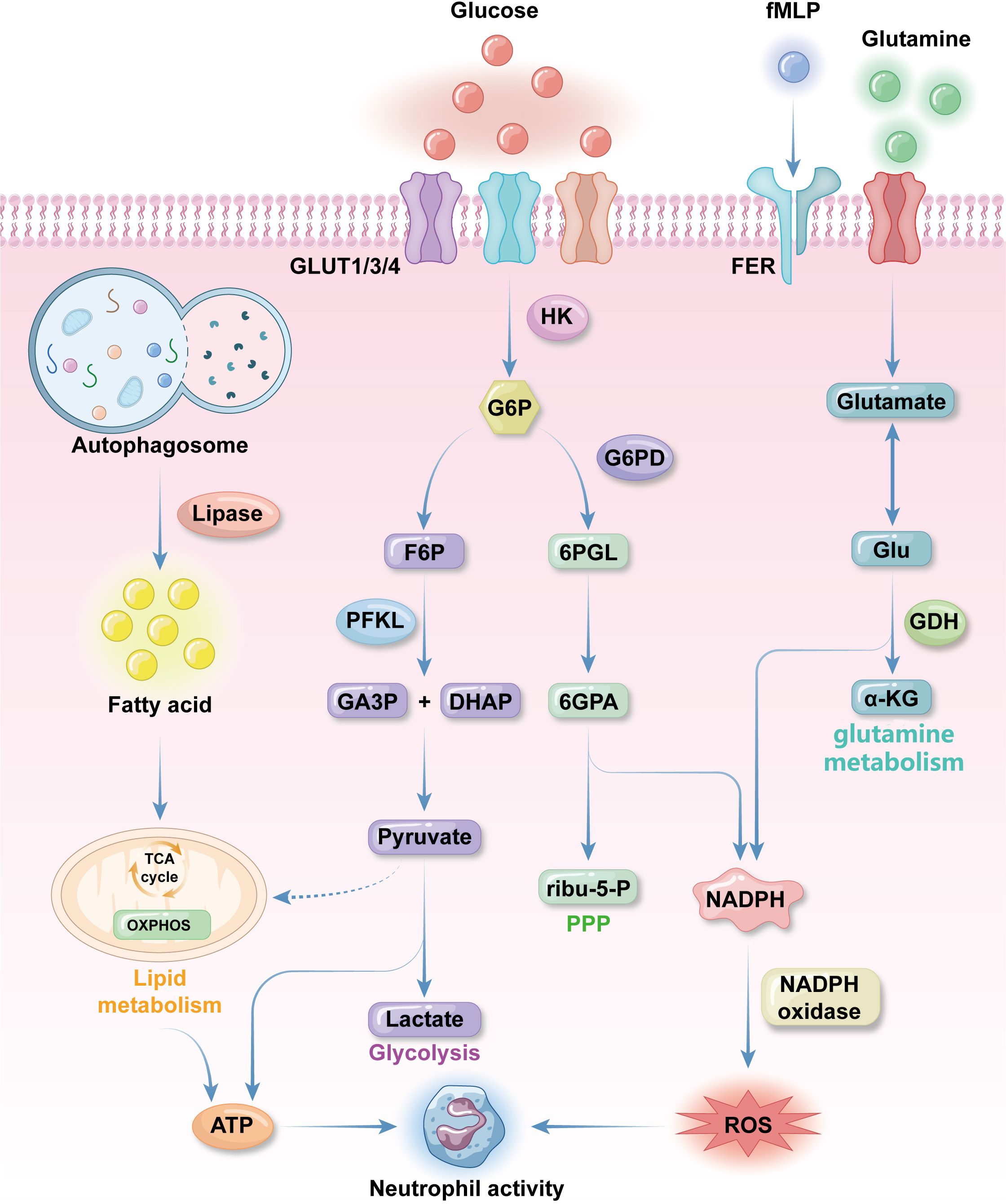
Figure 1. Fundamental metabolic features of neutrophils. neutrophils uptake glucose by the effect of glucose transporter proteins GLUT1, GLUT3 and GLUT4 expressed on the plasma membrane. Glucose will be converted to glucose-6-phosphate (G6P) by hexokinase 2(HK2) as immediately as glucose enter neutrophilic cytosol. G6P will be turned into 3-phosphoglyceraldehyde(GA3P) and dihydroxyacetone phosphate(DHAP), pyruvate and lactate in sequence for ATP synthesis in glycolysis. When neutrophils are activated pentose phosphate pathway (PPP) will be activated. PPP get 6-phosphogluconolactone(6PGL) from G6P through G6P dehydrogenase and finally get NADPH, which produce reactive oxygen species (ROS) for anti-bacterial activities. Glutamine is an important supplementation source of ROS. It is turned into glutamate, which can be transformed into Glu and then α-KG to get NADPH. Glutamine uptake and utilization can be promoted by formyl peptide receptors (FPR) with N-formyl-L-methionyl-L-leucyl-phenylalanine (fMLP) to promote NADPH synthesis. Lipid metabolism is the major energy source during neutrophils differentiation. Autophagosome use lipase to degrade lipid into fatty acids (FAs), which then get into TCA cycle to produce energy for differentiation and other neutrophils activities. Conventional mitochondrial metabolism pathways like TCA cycle and oxidative phosphorylation (OXPHOS) only occur before neutrophils finish differentiation because mature neutrophils contain a low density of mitochondria with low activities.
In some contexts, for example inflammatory sites or tumor microenvironments, there is limited glucose available to neutrophils; therefore, to maintain glucose homeostasis, neutrophils also have robust capacity to store and utilize glycogen (33). Glycogen storage dysfunction or replenishment leads to metabolic exhaustion and impaired neutrophil function, resulting in deficient neutrophil-related immune responses (52). G6P, which bridges glycolysis and glycogenesis, is a key regulator of glucose and glycogen homeostasis (53). The glucose-6-phosphotase (G6Pase) complex, which localizes to the endoplasmic reticulum membrane, converts G6P to glucose, to maintain glucose and glycogen balance, rendering neutrophils the only leucocytes equipped with both gluconeogenesis and glycogenesis abilities (54). Glyceraldehyde-3-phosphate, an intermediate product of glycolysis, also regulates this process (55). Impaired glycogen synthesis from G6P leads to the autosomal-recessive syndrome, glycogen storage disease type Ib (GSD-1b), characterized by neutropenia and neutrophil dysfunction (56). GSD-1b is caused by deficiency in glucose-6-phosphate transporter (G6PT), as G6Pase-β function is closely associated with G6PT (57). The neutropenia observed in GSD-1b results from over-activated neutrophil apoptosis and abnormal neutrophil differentiation in the bone marrow, influenced by disordered energy supplementation and activation of transcription factors, including HIF-1α and PPAR-γ (58).
As mentioned above, activated neutrophils require ROS for their immunological functions. A major route to ROS production in activated neutrophils is through the pentose phosphate pathway (PPP) in the cytosol (59). Although glycolysis does not directly produce ROS, it does generate G6P for the PPP (60). Unlike the TCA cycle, the PPP utilizes G6P to efficiently synthesize large amounts of NADPH for ROS production, without generating any ATP (61, 62). Once neutrophils are stimulated, the PPP will be strongly triggered within 30 minutes in preparation for an immune response (63). G6P dehydrogenase is an important enzyme that catalyzes the conversion of G6P to 6-phosphogluconolactone, which initiates the PPP (61). G6PD deficiency is a genetic disorder characterized by susceptibility to infections and recurrent bacterial infections, due to insufficient NADPH and ROS production (64). Cytoplasmic ROS derived from the PPP is crucial in mediating the release of neutrophil extracellular traps (NETs), a process linked to both neutrophil immune effects and cell death (65) (Figure 1).
2.2 Mitochondria metabolismIn the quiescent state, neutrophils have low mitochondria density and weak mitochondrial activity, and the role of mitochondria in neutrophil function remains controversial (66, 67). Mitochondrial respiratory chain inhibitors, such as FCCP or oligomycin, which deplete mitochondrial membrane potential and depress mitochondrial ATP synthesis, respectively, do not decrease ATP production or basal oxygen consumption in neutrophils (68, 69). Further, temporary treatment using mitochondrial respiratory chain inhibitors has no influence on ROS production; however, prolonged mitochondrial suppression using oligomycin (incubation > 2 h) results in reduced ability to trigger respiratory burst (69). A metabolic shift from OXPHOS to glycolysis occurs during neutrophil differentiation in the bone marrow, and OXPHOS is a primary source of mitochondrial ROS (mROS) in many cell types, distinct from the cytosolic ROS synthesized by membrane receptors, such as formyl peptide receptors (FPR), during respiratory burst (70, 71). Bao et al. demonstrated that N-Formyl peptides expressed on invading bacteria (e.g., N-formyl-L-methionyl-L-leucyl-phenylalanine, fMLP) can stimulate neutrophilic respiratory burst by activating FPRs, and this interaction can be blocked by oligomycin (71). Subsequent studies clarified that ROS induced by fMLP originates from the cytosol and does not involve mROS production (72). It indicates that mitochondria may sense neutrophilic respiratory bursts, independently of OXPHOS (73) (Figure 1).
Mitochondria loss is also driven by the effects of HIF-1α on transcription during differentiation. Hypoxia activate HIF-1α to induce mitochondrial autophagy in neutrophils (50). Cytochrome c, a vital component of the electron transport chain, is downregulated on HIF-1α activation, leading to OXPHOS suppression (49). Another essential function of OXPHOS is creating membrane electrochemical potential (ΔΨm) across the mitochondrial membrane, which facilitates electron transfer and provides energy for ATP synthesis (74). Electron transfer is mediated by four mitochondrial respiratory chain complexes (CI, CII, CIII and CIV) in the inner mitochondrial membrane (75). The absence of cytochrome c results in loss of CI, CIII, and CIV within a respiratory chain, with consequent failure to produce sufficient energy for ATP synthesis (51, 75, 76). Although ΔΨm does not typically contribute to neutrophil energy supplementation and biosynthesis, diminished mitochondrial membrane potential can trigger neutrophil apoptosis (49, 69). To maintain cell viability, neutrophils utilize glycerol-3-phosphate to transport electrons. The ΔΨm generated with glycerol phosphate as a substrate is higher than that generated with other complexes, with or without substrate. Glycerol-3-phosphate can diffuse into the mitochondria immediately on its production during glycolysis, followed by oxidation to dihydroxyacetone-phosphate on the outer surface of the inner mitochondrial membrane. This process donates electrons to complex III of the respiratory chain via ubiquinol (46). The mROS inhibitor, MitoTEMPO, accumulates in mitochondria and reduces neutrophil superoxide production by inhibiting complex III, highlighting the critical role of complex III in mROS production (77). Inhibiting complex III also increases lactate production, as it prevents glycerol-3-phosphate from entering mitochondria. This process is considered to regulate aerobic glycolysis, without affecting ATP synthesis (46) (Figure 1).
Fatty acid oxidation (FAO) is another important feature of neutrophil mitochondrial metabolism in ATP-demanding situations, such as differentiation or tumor adaptation (78, 79). Concerning the unique role of lipid metabolism in neutrophil, it will be discussed in the next part.
2.3 Lipid metabolismIn recent years, there has been an increasing focus on lipid metabolism as an additional energy source, alongside glycolysis, in various neutrophil functions (36). Lipid metabolism serves as an important contributor to energy provision, cell signal transduction, and immune response regulation, under both physiological and pathological conditions.
Fatty acids (FAs) are essential energy sources for neutrophils, particularly when glucose is limited (79). Autophagy is the main producer of FAs in neutrophils through lipophagy, which supplies the majority of energy for neutrophil differentiation (78).During differentiation, lipid can be degraded into FAs, which is then converted into TCA cycles to synthesize ATP and provide most energy for cellular processes of differentiating neutrophils. PPARγ is a transcription factor that regulate adipogenesis and promote lipid droplet formation (80). G-CSF can stimulate the accumulation of LDs in neutrophils and accelerate the maturation of neutrophils through PPARγ (81).During infection, Platelet activating factor, LPS stimulation and cytokines like IL-5 facilitate LDs accumulation with signal platform of TLR4, TLR7 and TLR9 to enhance the immune response (82, 83). Lipophagy involves the transport of lipid droplets, which are membrane-bound lipid-storing particles in the cytosol, into double membrane-bound autolipophagosome vesicles, which are then delivered to lysosomes for oxidation into FAs (84). These FAs are then converted into fatty acyl-CoA esters, which are transported into mitochondria via an acetylcarnitine transporter. Once inside the mitochondria, fatty acyl-CoA esters are metabolized into acetyl-CoA molecules, which enter the TCA cycle and the OXPHOS system to generate ATP and NADPH (85). Neutrophil maturation and differentiation are predominantly regulated by autophagy related protein 7 (ATG7) in mice, deficiency of which disrupts lipophagy, hindering neutrophil differentiation and causing accumulation of immature neutrophils in the bone marrow (78). Besides autophagy, FAs can also be derived from the decomposition of excess lipids by neutral lipases, such as adipose triglyceride lipase and hormone-sensitive lipase, which help cover energy shortages under metabolic stress (86, 87) (Figure 1).
In inflammatory pathways, lipids serve as both independent signaling molecules in cell signal transduction, and as activators or modulators of certain effector proteins or transcription factors (88, 89). Lipid inflammatory mediators can be classified into three types according to their structures: arachidonic acid-derived eicosanoids, membrane phospholipids, and omega-3/6 FA-derived lipids (36). These molecules are vital regulators of proper immune responses, as myeloid-specific adipose triglyceride lipase-deficient mice, which fail to synthesize lipid mediators, exhibit disordered immunological features (90). Fatty acid transporters and fatty acid recognition receptors on neutrophils can also efficiently modulate inflammatory pathways by interacting with specific lipids in the immunological microenvironment to enhance innate immune responses, or promoting neutrophil recruitment to inflammatory sites (91, 92).
2.4 Glutamine metabolismGlutamine is an amino acid which remarkably modulates immune function in the human body. Neutrophils are among the immune cells that utilize glutamine and consume it at a higher rate than other leukocytes (93) (94). Anti-infection function of neutrophils can be facilitated by glutamine without influencing their phagocytic ability (95). This effect of glutamine on neutrophil function likely mediated by increased ROS production, as glutamine can generate NADPH through malate synthesis and activated NADPH oxidase, a key enzyme in ROS production, by upregulating the expression of its components, gp91, p22, and p47 (96) (Figure 1).
Deficient glutamine utilization or inadequate glutamine supply is strongly associated with various diseases, including cancers, diabetes, and sepsis, which may result from oxidative stress (97–99). Glutathione/γ-l-glutamyl-l-cysteinyl-glycine (GSH), the most abundant non-enzymatic antioxidant in human cells, is primarily distributed in the cytoplasm, and can directly react with ROS to eliminate peroxides (100). Oxidized GSH (GSSG) is produced during this reaction, thus cell redox state can be reflected by the GSH/GSSG ratio (101). Glutamine, in the form of glutamate, is a crucial precursor amino acid for GSH and has a vital role in GSH synthesis; hence, glutamine levels determine the GSH/GSSG ratio by modulating GSH synthesis, which facilitates neutrophil function under oxidative stress (98).
3 Metabolic alterations in different types of neutrophil death3.1 ApoptosisApoptosis is among the most common forms of cell death in neutrophils under physiological conditions. Neutrophil apoptosis usually results from caspase activation through various pathways, and is characterized by changes in cell shape, pseudopod retraction, volume reduction, and chromatin condensation (102–104), and observation of these specialized changes under a microscope indicate that apoptosis is occurring.
Neutrophil apoptosis initiation can be divided into two main pathways, intrinsic and extrinsic. The extrinsic pathway involves activation of death receptors and members of the tumor necrosis factor receptor family, distributed on the plasma membrane (105, 106). Death receptor pathways, such as CD95 (Fas/APO-1), can respond to exogenous and endogenous ROS in neutrophils, inducing apoptosis (107). The intrinsic pathway is triggered by cytochrome c release from mitochondria into the cytosol, regulated by the bcl-2 family of apoptotic proteins, which maintain mitochondrial integrity (108). Cytochrome c interacts with the cytosolic protein, apoptotic protease-activating factor 1 (Apaf-1), to activate the effector, caspase-9 (109). Further, cytochrome c is released as the outer mitochondrial membrane is destroyed, due to loss of mitochondrial membrane potential, which can be diminished by treatment with mitochondrial respiration inhibitors, such as oligomycin, to initiate apoptosis even before apoptotic morphological features appear (69). Nevertheless, inhibiting mitochondrial function does not accelerate neutrophil apoptosis (69).
Glycolysis is inhibited in apoptotic neutrophils, which cannot trigger respiratory bursts, leading to decreased pathogen killing ability (110, 111). As previously mentioned, mitochondrial membrane maintenance in neutrophils is fueled by glycerol-3-phosphate produced during glycolysis, suggesting that reduced glycolysis may trigger neutrophil apoptosis (46). Limited glucose (0.6 mM) is a key promoter of neutrophil apoptosis, an effect enhanced by G6Pase-β deficiency in patients with GSD-Ib (112). G-CSF, which is used to treat neutropenia in various conditions, can delay, but not prevent, neutrophil apoptosis by repairing glucose uptake and utilization (112). Unlike in other cells, hypoxia delays apoptosis in neutrophils, both by reducing the ROS levels crucial for apoptosis, and by activating HIF family transcriptional factors (107, 113–115). HIF-1α primarily modulates neutrophil metabolism for adaptation to anoxic environments (114). Li et al. demonstrated that cyclosporine can inhibit the SIRT6-HIF-1α-glycolysis axis to accelerate neutrophil apoptosis (116). In contrast, HIF-2α activity directly promotes the intrinsic apoptosis pathway (115). These mechanisms help prolong the lifespan of neutrophils and reinforce immune function in low-oxygen sites of inflammation (117) (Figure 2).
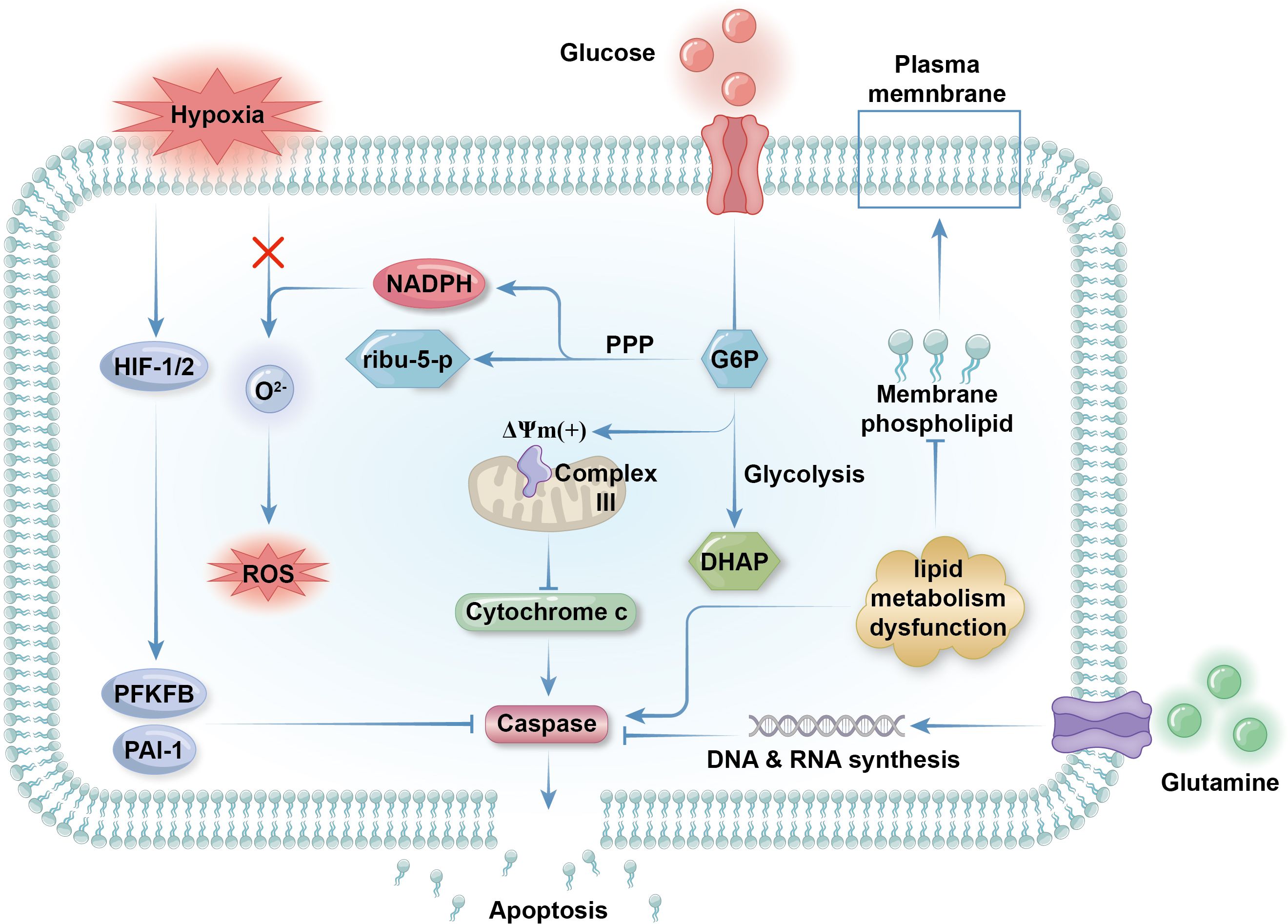
Figure 2. Metabolic regulation of apoptosis. Glycolysis is inhibited as neutrophilic apoptosis is triggered. Glycolysis contributes membrane electrochemical potential (DYm) to respiratory chain complex III expressed on the outer membrane of mitochondria to maintain the integrity of the organelles. If glycolysis is low, the mitochondria will rupture and release cytochrome c into the cytosol, which activates apoptotic effector caspase and initiate the programmed cell death. Hypoxia delay apoptosis by activate transcriptional factors HIF1 and HIF2 as the promote the expression of anti-apoptotic protein like PAI-1 and 6-phosphofructo-2-kinase (PFKFB). Concerning that ROS can initiate apoptosis by activating death receptors like CD95 on plasma membrane, preventing ROS production by limiting peroxide like O22- is another way of hypoxia rejecting apoptosis. Although lipid metabolism is in a low state in mature neutrophils, impaired lipid metabolism that inhibit the synthesis of membrane phospholipid can result in apoptosis as well. Glutamine metabolism get weaker during apoptosis because of decreased demand for NADPH in apoptotic neutrophils. Surviving cells need abundant glutamine supply for the synthesis of nucleic acid for the transcription and expression of anti-apoptotic genes.
Glutamine metabolism is crucial for NADPH generation and is downregulated in apoptotic neutrophils, due to reduced NADPH demand. Besides boosting respiratory bursts, glutamine also produces precursor nucleotides for RNA and DNA synthesis, and is responsible for expression of surface activation proteins and cytokine production (118). Glutamine supplementation can effectively slow neutrophil apoptosis, particularly exercise-induced apoptosis, and modulates immunological functions (37, 119). While glutamine fuels ROS generation, it suppresses extrinsic apoptosis by decreasing p38, MAPK, and JNK phosphorylation and reducing p53 and caspase-3 expression (120). Interestingly, glutamine extends the life span of neutrophils and enhances their function while exhibiting anti-inflammatory effects, possibly due to its protective effects on other cells (121) (Figure 2).
The effects of lipid metabolism regulation on neutrophil lifespan are variable. Neutrophils barely utilize FAO at the end of their lives, when their energy needs are markedly reduced. Dysfunction of lipid metabolism can suppress peroxisome-derived membrane phospholipid synthesis, leading to neutrophil apoptosis without affecting neutrophil energy metabolism or differentiation (122). Inflammatory mediators derived from lipolysis are important in propelling neutrophil apoptosis, mainly through death receptor signaling pathways (102). For example, 15-deoxy (Delta)12-14PGJ2, a cyclopentenone prostaglandin derived from arachidonic acid, induces neutrophil apoptosis in rats, possibly through PPARγ inactivation (123, 124). Phospholipolysis is a vital source of arachidonic acid in neutrophils, and phospholipase and sphingomyelinase activity, which produce endogenous lipid mediators from membrane phospholipids, are closely associated with normal apoptosis progression (125). Overall, lipid metabolism can have dual effects on neutrophil survival, and the purpose of lipolysis and lipogenesis in neutrophils varies considerably in the contexts of different diseases (Figure 2).
Neutrophil apoptosis can be caused by extracellular stimulation or damage, as well as by scarcity of anti-apoptotic molecules, including anti-apoptotic proteins and pro-inflammatory signaling molecules (36, 126). Anti-apoptotic proteins, for example, the bcl-2 family member, Mcl-2, are highly expressed in neutrophil cytosol and their levels decrease before other apoptotic changes occur (126). As Mcl-2 has a shorter half-life than neutrophils (around 2–3 h) and neutrophils do not express other bcl-2 family proteins, vigorous protein synthesis is required for neutrophil survival, and impaired protein and amino acid metabolism can lead to apoptosis (Figure 2).
NETosis is a specialized form of cell death in neutrophils that shares many mechanistic features with apoptosis. NETosis and apoptosis appear to be two sides of the same coin, with various factors determining which pathway a dying cell will take. The mechanisms, similarities, and differences between NETosis and apoptosis are discussed in the following section.
3.2 NETosisNETs are web-like chromatin structures released by neutrophils that are important for neutrophil immune responses and pathogen elimination (65). Accompanied by nucleus disintegration, cell membrane collapse and cell dysfunction, NET formation and release are features of a unique type of cell death, differing from apoptosis and necrosis, NETosis (127, 128). However, in some cases, neutrophils can maintain biological and immunological function after releasing NETs (129). Hence, NETosis is categorized according to its effect on neutrophil viability as suicidal and vital NETosis (129). Although suicidal and vital NETosis share notable common mechanisms among their key steps, including NET assembly and release, they lead to vastly different outcomes for neutrophils. Therefore, the Cell Death Nomenclature Committee does not recommend using the term NETosis to describe the vital process of NET release (130). In this review, we focus solely on the metabolic mechanisms that are either shared between vital and suicidal NETosis or exclusive to suicidal NETosis.
Neutrophil activation is required to trigger NETosis; hence, the metabolic features of NETosis initiation are aligned with those of neutrophil activation, including upregulation of glycolysis, lipid metabolism, and respiratory burst (34, 131). Neutrophils can produce ROS through NADPH oxidase activity and utilize it for NETosis (132). NETosis and apoptosis are generally considered mutually exclusive cell death processes that contribute to opposing metabolic alterations. Although ROS can activate both NETosis and apoptosis, the Akt and JNK pathways mediate the switch from apoptosis to NETosis in a ROS production-dependent manner (133, 134). Phorbol 12-myristate 13-acetate (PMA), a widely used NETosis activator, enhances the activity of NADPH oxidase 2 (NOX2), leading to synthesis of large amounts of cytosolic ROS, thus triggering Akt pathway to promote NETosis and inhibit apoptosis (134). Akt can also be activated to initiate NET release, as sensor kinases in the JNK pathway are triggered on stimulation by gram-negative bacteria or high levels of lipopolysaccharide (133). Meanwhile, recent evidence indicates that, in some specific cases, such as ultraviolet stimulation, another type of cell death, aponetosis, with features of both NETosis and apoptosis, is triggered through an NADPH oxidase-independent pathway; the metabolic features of aponetosis require further investigation (135) (Figure 3).
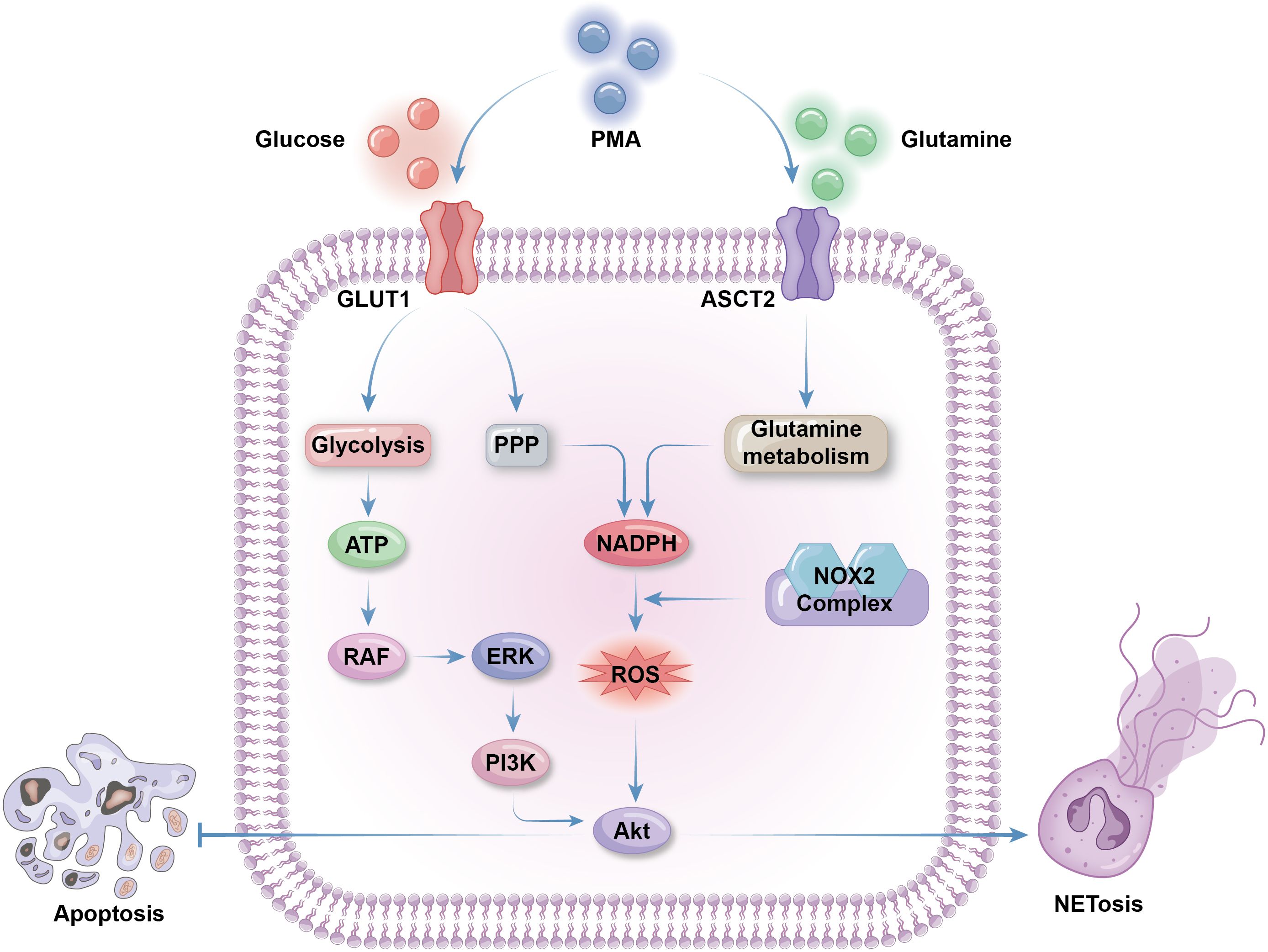
Figure 3. Metabolic selection of apoptosis or NETosis. The initiation of NETosis greatly depends on glucose metabolism, including glycolysis to produce ATP and PPP to produce NADPH. Glycolysis activate RAF via ATP production. RAF then activate ERK to trigger PI3K/AKT pathway to promote NETosis and inhibit apoptosis. Cytosolic ROS produced by NADPH oxidases 2(NOX2) is necessary for AKT production. Besides of enhanced PPP activity, glutamine metabolism is also facilitated to supply extra NAPDH for NETosis. Both glucose and glutamine uptake and metabolism can be enhanced by NETosis activator phorbol 12-myristate 13-acetate (PMA) by promoting the expression of membrane glucose transporter GLUT1 and membrane glutamine transporter ASCT2.
Abundant glucose is indispensable for NETosis, and glycolysis is the main glucose metabolic pathway that fuels NETosis. In the context of diseases associated with overactivated NETosis, such as cystic fibrosis or asthma, neutrophils from airway connective tissue contain increased levels of glycolytic metabolites and exhibit enhanced glucose uptake (136, 137). 2-deoxy-D-glucose, a glucose analogue that competitively binds to the glycolytic rate-limiting enzyme, glucose hexokinase, blocks NETosis by inhibiting glycolysis (138). NET formation can be divided into two phases: the first phase is chromatin decondensation, which does not rely on glycolysis; while the second is glycolysis-dependent NET release. Activated neutrophils grown on glucose-free media undergo morphological changes to their nuclei, without release of NET vesicles (138). PMA-stimulated NETosis and NETosis in cystic fibrosis are accompanied by upregulated expression of GLUT1, rather than GLUT3 or GLUT4, which is a primary contributor to enhanced glucose metabolism (138, 139). As discussed above, PPP is the most important source of ROS production in neutrophils, producing ample NADPH for NADPH oxidase activity. The downstream glycolytic enzyme, phosphofructokinase liver type (PFKL), converts fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate, preventing F6P entry into the PPP, thus blocking NADPH and ROS production (140). Hence, proper PPP initiation is necessary for ROS supply for NETosis, despite enhanced glycolysis (141) (Figure 3).
There is a weak association between lipid metabolism and NETosis levels under physiological conditions, as NETosis usually occurs in mature neutrophils with little reliance on lipid metabolism for energy production (102). However, in some diseases, NETosis can be abnormally activated through certain specialized lipid metabolism pathways. LNK is a member of the SH2B family of adaptor proteins that acts as a key regulator of hematopoietic cell signaling and proliferation (142, 143). Together, LNK deficiency and hyperlipidemia lead to platelet production and activation, platelet–neutrophil aggregates, and neutropenia caused by overwhelming NETosis, resulting in vascular or coagulation disorders (144, 145). LNK dysfunction promotes neutrophil priming and response to oxidized phospholipids from activated platelets, accelerating NETosis. This process can be completely blocked by deficiency of peptidyl arginine deiminase 4 (PAD4), a key effector of DNA decondensation (145). Disruption of wound healing and cardiovascular disorder in patients with diabetes or model mice may also be a consequence of NETosis mediated by abnormal lipid metabolism and PAD4 function (146, 147). Insulin resistance promotes fatty acid uptake and oxidation, leading to increased ROS production in neutrophils. Excessive ROS stimulates the function of transcription factors, such as PPARα, and the expression of enzymes, including PAD4, which help to trigger NETosis (148, 149). During the development of atherogenesis, cholesterol facilitates neutrophil recruitment and NET formation, which is mediated by activation of the proinflammatory NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome protein (150). Unusual lipid metabolism in various cells, such as cholesterol accumulation in macrophages within atherosclerotic plaques, can also stimulate NETosis, due to excessive secretion of IL-1β by these macrophages (151) (Figure 3).
Glutamine is crucial for NADPH oxidase activity and ROS production, making it significant for NETosis initiation. PMA stimulates expression of the NADPH oxidase components, gp91, p22, and p47, to facilitate NET release, which can be blocked by the glutamine metabolism inhibitor, 6-Diazo-5-oxo-l-norleucine (96). Glutamine depletion partially, but not completely, diminishes NET formation, as glucose is the primary source of ROS for most neutrophilic antibacterial activities (138). Macrophages use glutamine to synthesize nitric oxide (NO) from arginine through the inducible NO synthase enzyme, utilizing NADPH as an energy source; however, there is minimal evidence supporting this pathway in neutrophils (Figure 3).
3.3 Necrosis and necroptosisNecrosis is a form of cell death that is less regulated than apoptosis, which is initiated in response to detrimental environmental factors, such as lack of oxygen or essential nutrients, toxic substances, or extremely acidic or basic conditions (103). Similar to apoptosis, necrosis involves changes in the shape of cytoplasmic organelles, chromatin condensation, and oligonucleosomal DNA fragmentation, but without caspase activation (152). Necroptosis is a type of programmed cell death that shares more features with necrosis than apoptosis, and is primarily regulated by receptor-interacting protein kinase-3 (RIPK3) and mixed lineage kinase domain-like protein (MLKL) (153, 154). When apoptosis is blocked, for example, due to bcl-2 family protein dysfunction, aging or apoptotic neutrophils may undergo cell clearance via necroptosis (155).
Although necrosis and necroptosis are typically independent of the caspase cascade, ROS and Ca2+ ions are key participants that do not directly relate to external stimuli in these two processes. During necrosis, excessive Ca2+ inflow causes mitochondrial calcium overload, and ROS destroys lipid, protein, and DNA structures, which jointly lead to disordered ionic equilibrium and mitochondrial dysfunction. In necrosis, unlike apoptosis, mitochondria usually swell, which greatly impairs OXPHOS and ATP generation. Consequently, apoptosis is blocked due to deficient release of apoptotic proteins from the mitochondria under remote energy supply conditions (156). In necroptosis, ROS generation largely depends on mitochondria, using a mechanism involving mitochondrial permeability transition related to cyclophilin D, rather than Bax or Bak (155). However, necroptosis can be triggered normally in cells with a low density of mitochondria, such as neutrophils (157).
3.4 AutophagyAutophagy is an important cellular process that maintains metabolic equilibrium when energy or nutrients are deficient. In neutrophils, autophagy is indispensable for immune response regulation and pathogen elimination, as neutrophils are often present in environments lacking sufficient oxygen or energy sources. Autophagy can also be responsible for removal of harmful substances, such as damaged mitochondria and protein aggregates in the cytosol, a process some researchers refer to as selective autophagy (158). In addition to selective autophagy, macroautophagy and microautophagy are processes triggered by distinct pathways that result in similar cellular consequences (159).
Autophagy is a form of cell death used to compensate for energy deficiency and inhibits other cell death processes, such as apoptosis or NETosis. For example, dysfunction of PD-L1, which promotes neutrophil autophagy, results in failure of NET formation and release (160). In cases where NETosis is activated by PMA stimulation and energy substrates, such as glucose or glutamine, are relatively restricted, autophagy can be initiated to provide energy for NET release and inhibit apoptosis (161). Under stressful situations, for example, limited glucose, selective autophagy can degrade specific proteins via lysosomes for nutrient and energy supplementation. This process, regulated by metabolic status, is important for maintaining metabolic balance (162). However, this type of autophagy can also consume excessive metabolic proteins, such as hexokinase 2 (HK2), impairing normal cellular metabolism and triggering unexpected cell death (163). Dysregulated selective autophagy primarily affects glucose and lipid metabolism, while its specific effects on neutrophils and neutrophilic metabolism remain elusive (164).
Autophagy is an essential source of FAs for neutrophils. As outlined earlier, autophagy mediates lipolysis to generate FAs for mitochondrial respiration, which provides energy for neutrophil differentiation in the bone marrow autophagy mediates lipolysis to degrade lipid and generate FAs for mitochondrial respiration, which provides energy for neutrophil differentiation in the bone marrow through FAs oxidation (78). Autophagy deficiency inhibits neutrophil degranulation by decreasing ROS production from fatty acid-mediated NAPDH oxidase activity (165). FAs or lipids can also regulate neutrophil autophagy (166, 167). He et al. revealed the role of β-hydroxybutyrate (BHB) in regulating neutrophil autophagy in cows (167). Treatment with BHB facilitates neutrophil adhesion by preventing the degradation of adherent molecules, such as CD11a, CD11b, and CD18, rather than increasing their expression levels. BHB also increases the mRNA abundance and production of the pro-inflammatory factors, IL-1β, IL-6, and TNF in bovine neutrophils, collectively inhibiting neutrophil autophagy (167).
3.5 PyroptosisPyroptosis is a newly discovered type of cell death that primarily relies on the activity of caspase-1 and is distinct from other forms of cell death, such as apoptosis. Neutrophil pyroptosis primarily involves processing of specific inflammatory cytokines via the inflammasome to modulate an appropriate immune response, and is usually initiated by intracellular bacterial infections; for example, Salmonella carrying Salmonella pathogenicity island 1 type III secretion systems (168, 169). Salmonella infection boosts assembly and function of the inflammasome, a protein complex that modifies precursors of the inflammatory cytokines, IL-1β and IL-18, driving pyroptosis (168). Activated NLRP3 and NLRC4 inflammasomes promote cleavage of the protein, gasdermin D (GSDMD), and its functional N-terminal proteolytic fragment induces neutrophils to undergo pyroptosis and produce IL-1β (170). In addition to killing intracellular pathogens, pyroptosis is involved in neutrophil secretion of IL-1β and IL-18. Further, in some situations, such as during acute Salmonella infection, neutrophils can escape cell death and maintain IL-1β production by activating the NLRC4 inflammasome (171).
Canonical inflammasome activation by Salmonella infection also facilitates NETosis and triggers pyroptosis through the NLRP1/3-GSDMD axis (172, 173). NOX2/gp41 deficiency does not block, but rather fosters, compensatory NET release, while GSDMD is unnecessary for PMA-induced NETosis, which largely relies on NAPDH oxidase activity (172). Although mROS is an established NLRP3 inflammasome activator, insufficient neutrophil mitochondrial function limits ROS activity as a powerful regulator of neutrophilic pyroptosis (174, 175). Circulating mitochondrial DNA can act as an effector to activate the canonical inflammasome, and is released on stimulation by calcium influx, along with mROS or other pyroptosis-inducing factors (176, 177). While mitochondria have diverse roles in regulating pyroptosis, there is limited evidence of their significance in neutrophils, which contain low numbers of mitochondria.
In dendritic cells, promotion of pyroptosis involves switching of the major metabolic pathway from OXPHOS to glycolysis, similar to the neutrophilic maturation process (178). Along with other chemical and biological inhibitors of hexokinase, N-acetylglucosamine, a component of the bacterial cell wall, mediates the relocalization of hexokinase from the surface of mitochondria to the cytosol, driving the NLRP3 inflammasome pathway and pyroptosis, independent of mROS and mitochondrial DNA (179).
Upregulated fatty acid supply or synthesis can facilitate inflammation through inflammasome activation (180). In contrast, limited fatty acid availability leads to reduced ROS production and increased autophagy, due to the anti-inflammatory effect of AMP-activated protein kinase, thereby inhibiting other forms of cell death and the inflammatory response (181). However, when glucose is scarce, FAO is abnormally activated to produce sufficient energy. During this process, ketone bodies such as BHB are produced, which can inhibit NLRP3 inflammasome activation through various pathways (182). Butyrate, a short-chain fatty acid with structural similarity to BHB, also has an anti-inflammatory effect by silencing the inflammasome (182). Indeed, short-chain FAs produced by gut microbiota, which are engaged in lipid metabolism in most cell types, have shown remarkable anti-inflammatory effects through various mechanisms, including control of the inflammasome and pyroptosis (183).
3.6 FerroptosisFerroptosis is an emerging form of cell death, with some features of necrosis, that has become an increasing focus of attention since it was first described in 2012 (184). Peroxidation of phospholipids mediated by intracellular iron accumulation is the most important characteristic of ferroptosis, which is regulated by a series of cellular and molecular mechanisms. While the roles and mechanisms of ferroptosis in cancer and ischemic diseases have been widely studied, its impact on neutrophil function is poorly understood (185). Nevertheless, several studies have explored the impact of neutrophil ferroptosis on modulating the tumor microenvironment, revealing that it can have both anti- and pro-tumor effects, depending on the context (14, 19, 186).
Lipid metabolism is the most pivotal metabolic pathway involved in ferroptosis across various cell types. Accumulation of cellular lipid peroxides drives ferroptosis; the peroxides are mainly derived from polyunsaturated FAs (PUFAs), such as arachidonic acid, which make up membrane phospholipids (187). The GSH-dependent lipid hydroperoxidase, GSH peroxidase 4 (GPX4), is a crucial regulator of ferroptosis, that can reduce phospholipid hydroperoxides through GSH oxidation, thereby preventing ferroptosis (188). In addition to lipid hydroperoxidases, several endogenous mechanisms eliminate lipid peroxides or prevent their production. Lipophilic radical trapping antioxidants (RTAs), such as tetrahydrobiopterin or coenzyme Q10, are endogenous reductive products that inhibit ferroptosis (189, 190). Endogenous RTA synthesis is triggered by ferroptosis suppressor protein 1 (FSP1), which is located on the plasma membrane, and FSP1 levels can be modulated by the antioxidant transcription factor, NRF2 (190, 191). During ferroptosis, general lipid metabolism, including PUFA metabolism, MUFA metabolism, and phospholipid synthesis and remodeling, is reprogrammed (192).
Although lipid oxidation is a central feature of neutrophilic ferroptosis, its metabolic characteristics, including glucose and glutamine metabolism, have only been explored in a limited number of studies. Glutamine metabolism is critical in neutrophils, and GSH serves as a major antioxidant, levels of which are closely related to cellular oxidative state and ROS production. Gao et al. and Xiao et al. reported that, in cancer cells, ferroptosis is controlled by glutamine metabolism and supply, as well as being regulated by cellular ROS levels (193, 194). Glutamate transporters, such as SLC7A11 and SLC1A5, modulate ferroptosis initiation by adjusting glutamine and GSH levels (195).
Glucose starvation limits neutrophil ROS production, suggesting that ferroptosis is proportionately linked to glucose metabolism. Moreover, energy depletion caused by glucose starvation activates the AMPK pathway, which blocks ferroptosis (196). As an energy-sensing kinase, AMPK is activated by energy insufficiency to inhibit the synthesis of polyunsaturated FAs that are indispensable for ferroptosis (197, 198). However, it remains unclear whether glycolysis or the PPP is the main glucose metabolic pathway during ferroptosis.
Similar to pyroptosis, ferroptosis is regulated by mitochondrial function, as mitochondria contribute substantially to glutamine metabolism, ROS synthesis, and lipid peroxide accumulation (199). although the role of mitochondria in neutrophil ferroptosis is elusive, due to continuous changes in mitochondrial activity and density in these cells.
4 Conclusion and futureStudying the metabolic features of immune cells is essential to understanding immune system function and regulation. In recent years, the metabonomics of innate immune cells, particularly neutrophils, has become the focus of increasing research, leading to elucidation of complex characteristics of the innate immune system. Although most innate immune cell types rely heavily on glucose metabolism, neutrophils, natural killer cells, and M2 macrophages predominantly utilize glycolysis, with almost no OXPHOS or respiratory electron transport chain activity (33, 200, 201). On neutrophil activation, the PPP is promoted, alongside glycolysis, to supply NADPH for antibacterial activity. Mitochondria are present at a low density in neutrophils and have altered function, barely participating in neutrophil energy metabolism; hence, the effects of mitochondria regulation on neutrophils are controversial (202). Lipid metabolism generally contributes to neutrophil differentiation and maturation, then decreases with increasing mitochondrial activity and OXPHOS. F
留言 (0)