
記住我



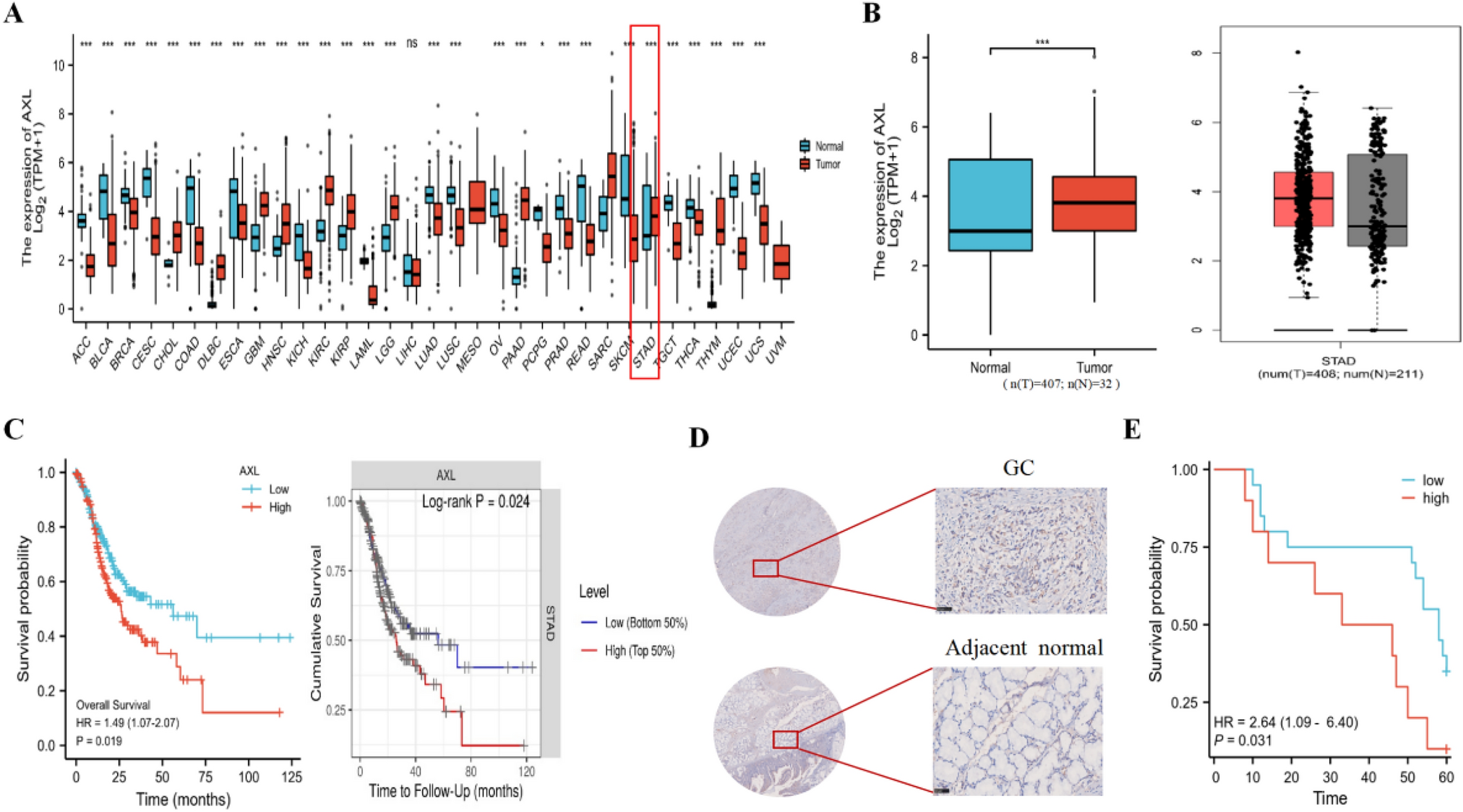
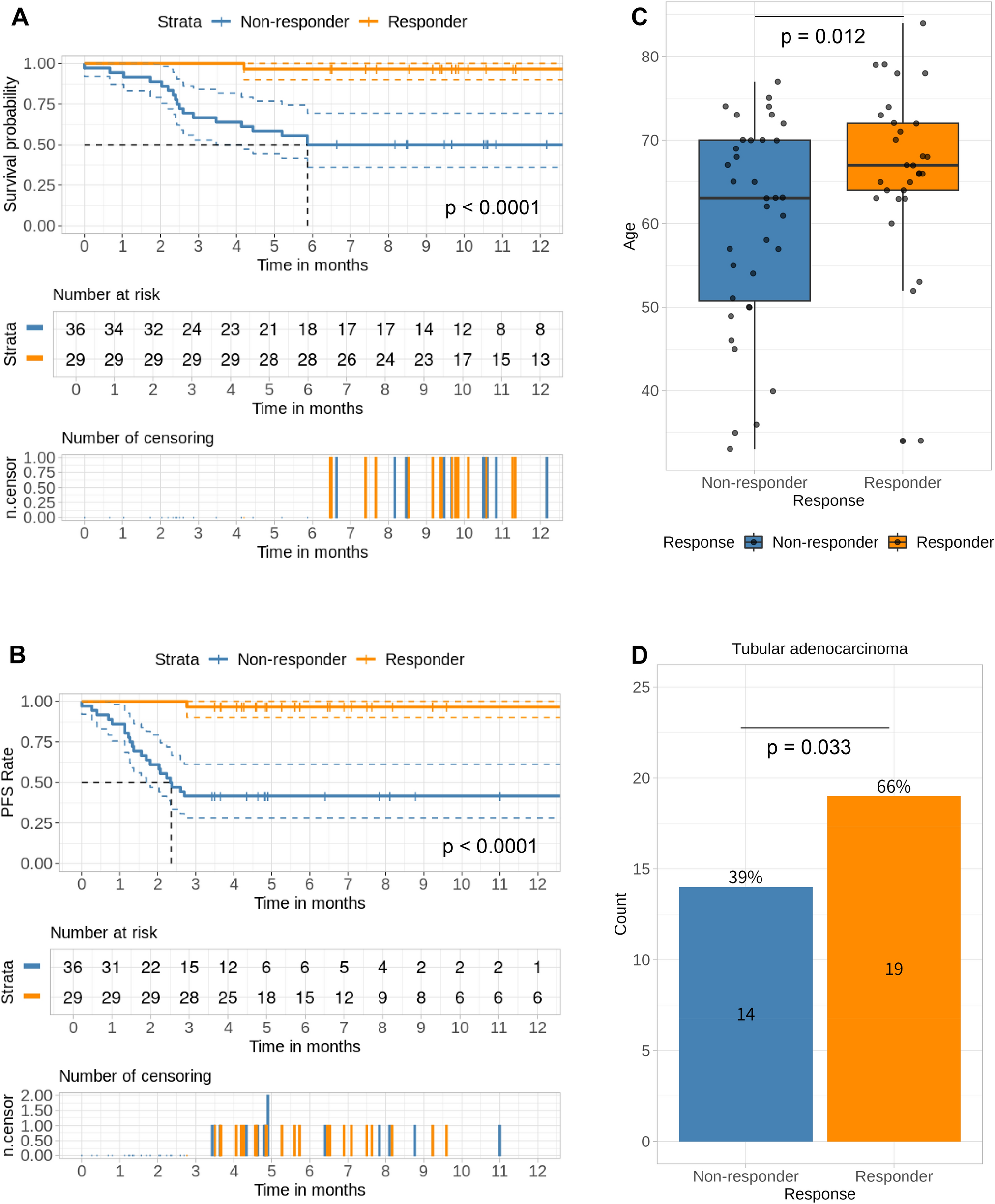





We conducted a comprehensive genomic and transcriptomic analysis of the tumor biopsy specimens before chemotherapy from 65 treatment-naïve unresectable or metastatic GC patients, which were enrolled in a translational study conducted at the National Cancer Center Hospital, Japan, from January 2013 to December 2017. Almost subjects (91%; 59/65) were treated with palliative chemotherapy containing fluoropyrimidine plus platinum agents. The details of the subjects are shown in Table 1. The patients who received chemotherapy were classified into five groups: FP (fluoropyrimidines + platinum), FP + Docetaxel, FP + HER2 (Trastuzumab), PTX (Paclitaxel), and others. Comparing the age (Supplementary Fig. 1A. P-value = 0.99 by ANOVA test), gender (Supplementary Fig. 1B. P-value = 0.63 by Pearson’s Chi-squared test), and disease status (Supplementary Fig. 1C. P-value = 0.86 by Pearson’s Chi-squared test) of patients who received chemotherapy, no significant differences were observed. Therefore, we considered the impact of treatment differences on the outcomes to be negligible. Of the 65 patients, the objective responses were as follows: Complete Response (CR) in 0 cases, Partial Response (PR) in 30 cases, Stable Disease (SD) in 16 cases, and Progressive Disease (PD) in 19 cases. Patients who showed PR were classified as 'Responder,' while those who showed SD or PD were classified as 'Non-responder.' Based on these classifications, we examined its association of clinico-pathological information (Table 1). Differences in response rates among different treatment regimens were also not observed (Supplementary Fig. 1D). The comparison of the 12-month survival period and progression-free survival (PFS) of Responders and Non-responders is shown in Fig. 1A and B. The responders showed better outcomes compared to Non-responders (Fig. 1A: P < 0.0001 by log rank test, and Fig. 1B: P < 0.0001 by log rank test). Furthermore, between Responders and Non-responders, significant differences were observed in their age (Fig. 1C) and histologic type (Fig. 1D, Supplementary Fig. 2A, and (Table 1), and trends were observed in cT:4a and cN:0 (Supplementary Fig. 2B and 2C, and Table 1).
Table 1 GC Patients characteristicsFig. 1
Clinical data from a cohort of 65 GC patients. A Overall survival (OS) and B Progression-free survival (PFS) in the two groups of chemotherapy Responders and Non-responders. C Comparison of age at diagnosis in the two groups of chemotherapy Responders and Non-responders. D Incidence of GC tissue type (Tub) in the two groups of chemotherapy Responders and Non-responders
Genomic and copy number profiles of 65 GCsDNA and RNA were extracted from fresh frozen core tumor biopsies from GC tissues before chemotherapy, and shallow whole-genome sequencing (× 1 ~ ; 65 cases) and RNA sequencing (65 cases) were performed. In addition, we made a panel of GC-related genes including 67 genes, microsatellite markers, drug-metabolizing genes, EBV and Helicobacter pylori (Supplementary Table 1), and targeted deep sequencing (65 cases) was also performed to detect GC-specific variants, pharmacogenomics (PGx) variants, and the pathogens. As a result, we detected 537 variants in the 56 genes (Supplementary Tables 2, 3). As shown in Supplementary Fig. 3A, somatic mutations were frequently found in several driver genes such as TP53 and MUC16; however, no difference in responsiveness to chemotherapy was observed in the presence of these variants (P > 0.05 Fisher’s exact test, Supplementary Fig. 3A, and Supplementary Table 1).
Considering the potential relevance of PGx variants to chemotherapy responsiveness due to their influence on individual drug metabolism, drug efficacy, and side effects, we evaluated six genes including GSTP1, DPYD, CYP2A6, TYMS, DPYS, and UGT1A1. We found that 32/65 (49.2%) of the GC samples had at least one PGx variant in the four genes excluding TYMS and DPYS (Supplementary Fig. 3B). However, no difference in responsiveness to the FP-based chemotherapy was observed (P > 0.05 Fisher’s exact test, Supplementary Fig. 3B, and Supplementary Table 1). Regarding to MSI status of 65 GCs, seven GCs showed MSI-high (Supplementary Fig. 3C), however, no difference in responsiveness to chemotherapy was observed in MSI/MSS (P > 0.05 Fisher’s exact test, Supplementary Table 1, and Supplementary Fig. 3C). EBV was detected with a read count ten times higher in three GCs (3/65; 4.6%) compared to other samples. Helicobacter pylori was detected in 29/65 (44.6%) of GC tissues, with ten cases showing more than tenfold the number of reads (Supplementary Fig. 3D). However, no difference in responsiveness to chemotherapy was observed in the presence of these pathogens (P > 0.05 Fisher’s exact test, Supplementary Table 1, and Supplementary Fig. 3D).
Subsequently, we analyzed somatic copy number alterations (CNAs) of GCs. Using genomic identification of significant targets in cancer (GISTIC) to identify statistically significant CNAs from the whole-genome sequencing data of 65 GCs, we detected copy number changes encompassing genes such as EGFR, MYC, CCND1, GATA6, CCNE1, and their surrounding regions, which have been reported to be associated with GC (Supplementary Fig. 4A). But no difference in responsiveness to chemotherapy was observed (P > 0.05 by t-test, Supplementary Fig. 4B). Next, we calculated the copy number signatures (CNS) for GC, which are patterns of somatic copy number variations (CNVs) that are characteristic of specific cancer types or subtypes. These signatures provide insights into how cancer cells accumulate genomic changes and the mechanical stress and DNA damage responses involved in the process [30], and we evaluated the CNS by using methods in the catalogue of somatic mutations in cancer (COSMIC) [30]. As a result, we extracted two types of CNS (GC_CNSignature1:Sig1 and GC_CNSignature2:Sig2) in our GC cohort (Supplementary Fig. 5A), and their comparison with the COSMIC signatures found a high similarity with COSMIC Signature CN1, CN4, or CN9 (Supplementary Fig. 5B). Importantly, a significant difference was observed in CN9 (= Sig1) between Responders and Non-responders (Supplementary Fig. 5C, P = 0.029 by Mann–Whitney U test). CN9 (= Sig1) has been reported to be positively associated with an increase in leukocyte fraction, elevated hypoxia scores, TP53 mutations, and PTEN mutations in specific cancers [34] [35] [36].
Differential expressions of immune-related genes is associated with chemotherapy responseIn RNA expression profiles of GCs from Responders and Non-responders, variability was observed in 492 genes; however, no specific pathways were identified using ReactomePA (Supplementary Figs. 6, and Supplementary Table 4). To investigate whether specific gene sets exhibited statistically significant expression changes between Responders and Non-responders, we conducted gene set enrichment analysis (GSEA). Utilizing the HALLMARK dataset and ImmuneScore (Fig 2A and B), we observed a significantly difference of the expression variations in the gene set below: HALLMARK_IL6_JAK_STAT3_SIGNALING (P = 0.045), HALLMARK_INFLAMMATORY_RESPONSE (P = 0.036) and HALLMARK_INTERFERON_GAMMA_RESPONSE (P = 0.039), ImmuneScoreESTIMATE (P = 0.018). Notably, many immune-related and inflammation-related signatures showed significant differences between Responder and Non-responders of GC.
Fig. 2
Multi-omics data from a cohort of 65 GC patients. A Comparison of the HALLMARK dataset between Responders and Non-responders. B Comparison of tumor microenvironment signature scores between Responders and Non-responders. C Immunological signatures, clinical data, and copy number signatures detected by targeted sequencing in 65 GC patients. These clusters are categorized based on the clustering of immune signatures
We previously reported that immune signatures and CNS differed between chemotherapy Responders and Non-responders in esophageal squamous cell carcinoma (ESCC) [37]. Therefore, we applied these approaches to this GC cohort. The CIBERSORT [38] deconvolution method, using the LM6 reference [39], classified these GCs into four immune subgroups (B cells, CD4 + T cells, CD8 + T cells, Neutrophils) (Fig. 2C). Interestingly, the Neutrophil group (P = 0.002) had more Responders, while the CD4 + T cells group (P = 0.001) had more Non-responders (Fig. 2C and Supplementary Fig. 7A). Clinical information showed no difference between Responders and Non-responders, nor among the respective subclusters (Fig. 2C). No significant changes were observed in the signatures of Neutrophils and CD4 + T cells based on histological type (P > 0.05, Supplementary Fig. 7B). Additionally, no significant relationship was found between various chemotherapy regimens and neutrophil signatures (P > 0.05, Supplementary Fig. 7C). Furthermore, one CNS CN9 (= Sig1) was significantly higher in the Neutrophil subgroup of immune signatures (Fig. 2B, and Supplementary Fig. 7D, P = 0.047 by Mann–Whitney U test). Comparing the 6-month survival rate (Supplementary Fig. 8A) and PFS (Supplementary Fig. 8B) of each immune subgroup, the Neutrophil subgroup had a better survival period compared to the other groups (Supplementary Fig. 8A: P = 0.044 by log rank test). These results suggest that immune signatures, neutrophil signatures, and CNS could influence the responsiveness of chemotherapy in GC.
Predicting the chemotherapy effectiveness by machine learning approachSundar et al. demonstrated that a Random Forest method could classify GC patients into Responders and Non-responders based on a 19-gene signature [40]. Interestingly, among the genes they identified, five (PTPRC, CD3G, TBX21, IL17A, FCGR3A) are associated with CD4+ T cells and neutrophils. Therefore, we hypothesized that several factors, including neutrophils, could have a complex impact on the efficacy of chemotherapy, and that a machine learning approach could be utilized to predict the effects of chemotherapy. To select a machine learning method, we initially performed 30 simulations by dividing integrated data with 123 of factors into 70% training and 30% test data using three methods: Random Forest, SVM (support vector machine), and Naïve Bayes, to measure accuracy. The results showed accuracies of Random Forest: 0.61, SVM: 0.59, Naïve Bayes: 0.46, leading us to choose Random Forest (Fig. 3A). To improve the predictive accuracy of the machine learning model, we aimed to select important features and eliminate irrelevant or redundant ones using the R package Boruta [41], evaluating the importance of all features (123 factors of genomics, RNA expression, signatures, and clinical information) through 10,000 simulations (Fig. 3B, Supplementary Fig. 8, Supplementary Table 5). As a result, immune clusters of neutrophils and CD4 + T cells, immune cell signatures of CD4 + T cells and B cells, INFLAMMATORY RESPONSE, and one CNS, CN9, were identified as important factors related to chemotherapy response in GC. Since the immune clusters reflect CIBERSORT scores, it was also suggested that the immune signature of neutrophils could be a key factor associated with chemotherapy response. Using these key factors as continuous variables, we constructed a diagnostic model to predict chemotherapy response using the Random Forest method. The predictive diagnostic model showed an accuracy of 0.80 and an AUC of 0.82 on the test data, with an AUC of 0.85 on the training set (Fig. 3C). Furthermore, we also ran this algorism on the data set of another cohort of GC patients (Ziyu Li et al. 2020 [33]. n = 35), which performed genomic and RNAseq of GC samples in China and analyzed the correlation of combination chemotherapy, and tested our predictive diagnostic model as validation dataset. The results for the Chinese cohort showed an AUC of 0.72 (Fig. 3D). Figure 3E illustrates the visualization of this diagnostic predictive model.
Fig. 3
Prediction of chemotherapy responsiveness in GC patients using machine learning approach. A Selection of machine learning models. We performed evaluations using Random Forest, SVM, and Naïve Bayes, and compared their accuracies. B Using the Boruta algorithm, six key factors were identified as important out of the 123 features. C The model was developed using 70% of the dataset and evaluated with the remaining 30% as the test data. AUC and ROC curve on the test data is shown. D AUC and ROC curve on the independent validation data from a Chinese GC cohort. E Random Forest, as an ensemble of multiple decision trees, was used to generate decision trees, and the tree most closely aligned with the Random Forest results was selected for further analysis
According to this model in Fig. 3E, neutrophils are the most critical factor to predict the response; a certain level of neutrophil presence in GC tissues increases chemotherapy sensitivity, while excessive infiltration leads to poor chemotherapy response (Fig. 3E, right branch). Also, the presence of B cells with minimal neutrophil infiltration predicted higher chemotherapy sensitivity. Conversely, a low presence of neutrophils with a high inflammatory response in cancer cells predicted poorer response. However, under these conditions, a lower presence of CD4 + T cells predicted better responsiveness. These results suggest that one reason for poor responsiveness in GC may be increased inflammatory response in the tumor microenvironment (TME) due to the presence of CD4+ T cells. Interestingly, the presence and balance of neutrophils were suggested to potentially influence chemotherapy responsiveness in GC.
Molecular characterization of neutrophil subclusters by single-cell RNAseqAs the next step, we focused on neutrophils in the tumor microenvironment (TME) of GC, which is one of the critical factors for predicting chemotherapy response in this study. Tumor-associated neutrophils (TANs) are a type of white blood cell present in the TME and play complex and diverse roles in cancer progression, metastasis, and immune response. The function of TANs in promoting or inhibiting tumor growth and metastasis depends on the TME. In our previous study, we reported that neutrophils infiltrating ESCC had a pro-tumor function that negatively impacted chemotherapy responsiveness [37]. In contrast, in this study, neutrophils infiltrating GC appeared to have an inhibitory function that positively affected chemotherapy responsiveness. By comparing this dual role, we hypothesized that characterizing neutrophils and other immune cells in TME could provide insights into their roles. We extracted CD45 + immune cells infiltrating tumor tissues from four ESCC patients [37] and four GC patients and performed sc-RNAseq. After filtering out low-quality cells and doublets (details in Methods), we annotated 59,584 immune cells infiltrating GC and ESCC using SingleR (Supplementary Fig. 9A).
Focusing on neutrophils, which are difficult to select and annotate using conventional methods [42], we used two approaches: a modified method by 10x Genomics and the method by Roudong Xue et al. [23]. As a result, we identified 12 subclusters of 3,822 neutrophils (Supplementary Fig. 9B), but Cluster 10 included clusters with a high frequency of immunoglobulins such as IGV3-1 (Supplementary Fig. 9C). Therefore, we excluded clusters or cells with a high frequency of immunoglobulins or those expressing suspicious lineage marker genes such as CD3E, CD19, NCAM1, CD8A, and CD4 (Supplementary Fig. 9E). We finalized these neutrophils formed from 12 subclusters, as shown in Fig. 4A. Figure 4B shows the distribution by GC or ESCC tissues and Supplementary Fig. 9E shows the distribution of anonymized samples. Analysis of the cancer type distribution in these subclusters revealed that Clusters 6 and 8 had a higher proportion of neutrophils infiltrating GC, while Cluster 10 had more neutrophils infiltrating ESCC (Fig. 4C).
Fig. 4
Single-cell RNA-seq analysis of neutrophils retrieved from GC and ESCC tissues. A t-SNE shows 12 sub-clusters of neutrophils retrieved from GC tissues (this study) and ESCC tissues (our previous study [37]). B t-SNE of neutrophils from ESCC and GC tissues, color-coded accordingly. C The number and percentage of immune cells from GC and ESCC tissues in each subclass. D The similarity between previously reported subclusters of neutrophils infiltrating the liver and those infiltrating gastrointestinal cancers. E Comparison of expression levels of gene markers in the clusters: TAN1 (CXCL8, CXCL1, CXCL2, ICAM1, CD44), TAN2 (HLA-DRA, CD74, HLA-DPB1), TAN3 (PLIN2, PLAU), TAN4 (RPL10, PRS2, RPS18, RPL3), and NETosis (PROK2, MME). F Annotation of neutrophils. G Stacked bar graph showing the distribution of annotated neutrophils in GC and ESCC. H Stacked bar graph categorizing neutrophils into Pro-tumor, Anti-tumor, and Other
Next, to characterize the 12 subclusters, we evaluated whether our cohort's neutrophil subclusters aligned with those reported in previous studies that documented the heterogeneity of neutrophils [42]. The heatmap in Fig. 4D shows the similarity between our cohort's neutrophil subclusters and those determined by the previous study [23]. Most clusters had a similarity of over 60%, suggesting that the neutrophils identified by SingleR and filtering were correctly selected. Interestingly, although many of the neutrophils infiltrating GC and ESCC showed similarities with those determined by the previous study [23], they were not distinctly separable as clusters and potentially exhibited complex combined characteristics (Fig. 4D). Moreover, Clusters 1, 2, and 6 were suggested to have unique features specific to GC or ESCC. Therefore, we considered it necessary to create a unique sub-classification of neutrophils for GC. Since classification markers for some TANs have been reported [24] [25] [16], we investigated genes related to these markers (Fig. 4E) and performed enrichment analysis using ReactomePA on genes specifically expressed in each cluster to identify pathways represented by each signature (Supplementary Fig. 10).
Clusters expressing TAN1 gene markers (CXCL1, CXCL2, CXCL8, ICAM1, CD44), associated with neutrophil activation, recruitment, cell adhesion, and NET formation, were found in Clusters 1, 3, 4, 5, and 11. Clusters strongly expressing TAN2 gene markers (HLA-DRA, CD74, HLA-DPB1, and other MHC II-related genes) associated with immunogenic antigen presentation and potential anti-tumor immunity were seen in Clusters 0, 6, 8, and 10. Clusters expressing TAN3 gene markers related to lipid metabolism (PLIN2 and PLPP3), which potentially play roles in activating extracellular matrix proteases and mediating tumor cell adhesion and movement through interaction with the homologous receptor uPAR expressed on tumor cells, were observed in Clusters 6, 1, 11, 5, 9, 3, 4, 10, and 8. Clusters expressing TAN4 gene markers (ribosomal-related genes RPL10, RPS2, RPS18, RPL3), suggesting a potential transition to tumor-associated neutrophils, were found in Clusters 6, 0, 2, 10, 7, and 8. NETosis gene markers PROK2 and MME were expressed in Clusters 1 and 11.
Based on these results, Cluster 0 showed high expression of immunoglobulin-related genes IGHA1 and IGKC, commonly expressed in B cells and plasma cells (Supplementary Table 6), and was involved in pathways related to protein synthesis and translation activation (Supplementary Fig. 10), suggesting characteristics of plasmacytoid neutrophils. Clusters 1, 3, 4, 5, and 11 (especially 1 and 11, involved in NETosis) displayed pro-tumor TAN characteristics. Clusters 2 and 7 contained pathways related to mitochondria and RHO signaling (Supplementary Fig. 10), suggesting that Clusters 2 and 7 may be activated neutrophils involved in promoting cell migration and motility (other pathways defined Cluster 2 as Migratory and Cluster 7 as Activated). Clusters 6 and 8 showed TAN2 markers and interleukin-related pathways, exhibiting anti-tumor TAN characteristics. Interestingly, Clusters 6 and 8 were prevalent in GC patients and, due to their clustering distance in Seurat dot plots (Fig. 4E), were suggested to be distinct types of neutrophils with different characteristics.
Cluster 9 had multiple pathways related to platelet activation, signaling and aggregation, thrombin signaling through proteinase-activated receptors (PARs), and thromboxane signaling through the TP receptor (Supplementary Fig. 10), suggesting pro-thrombotic neutrophil characteristics. Cluster 10, which included pathways involved in immune response regulation, cytokine signaling, and antiviral responses, had features of immunoregulatory neutrophils. These characteristics are summarized in Fig. 4F. The proportion of these annotated neutrophils in GC and ESCC (Fig. 4G) was analyzed, and the characteristics were grouped into pro-tumor, anti-tumor, and other categories, showing their proportions (Fig. 4H). The results indicated that GC contained a higher proportion of anti-tumor neutrophils, suggesting that the differences in neutrophil ratios could contribute to the chemotherapy responsiveness of GC and ESCC.
Neutrophil signature is related to chemotherapy response and survival of GCNext, we investigated whether these specific types of neutrophils are actually present in stomach tissue by using spatial transcriptome (ST) data of human gastric mucosa: stomach encyclopedia [26], and cell annotations were performed using the method described by Tsubosaka et al. [26]. From two ST datasets, we extracted 2,316 neutrophil fractions and created signatures for each of the neutrophil clusters identified in Fig. 4E using cluster-specific genes (Supplementary Table 7, and Supplementary Fig. 11). We then confirmed that these anti-tumor neutrophils were actually present within stomach tissues (Supplementary Fig. 12A). Additionally, we extracted neutrophil fractions from the ST data, identifying 463 pro-tumor neutrophils, 179 anti-tumor cluster 6 neutrophils, and other fractions, and displayed them using t-SNE (Supplementary Fig. 12B). However, Anti-Tumor Cluster 8 Neutrophils could not be confirmed due to the absence of detectable cluster-specific genes in the ST data.
After confirming the presence of Pro-tumor Neutrophils and Anti-Tumor Cluster 6 Neutrophils within stomach tissues, we created signatures using genes specifically expressed in these neutrophil groups (Fig. 5A, and Supplementary Table 8). Comparing these signatures within our GC cohort, we found that pro-tumor neutrophils showed significantly higher signals in Non-responders compared to Responders, while anti-tumor cluster 6 neutrophils showed significantly higher signals in Responders (Fig. 5B; pro-tumor neutrophils: P = 0.032, anti-tumor cluster 6 neutrophils: P = 0.034, Mann–Whitney test). Although data were limited and significant differences in survival and PFS could not be detected (Supplementary Fig. 13), these signatures were significantly associated with clinical outcomes in another clinical GC study [43] where only samples with available data for selected probes measured by the HGU133 plus 2.0 array were analyzed (Fig. 5C, N= 631; pro-tumor neutrophils signature: P = 6.6e-06, anti-tumor neutrophils signature: P = 2.3e-09). These results suggest that neutrophils with Pro-tumor or Anti-tumor characteristics may provide a synergistic benefit to patients, both alone and in combination with chemotherapy. Notably, in GC, the presence of a higher proportion of Anti-tumor neutrophils led to a greater number of patients in the immune cluster being classified as Responders. Conversely, in our previous study on ESCC, a higher proportion of Pro-tumor neutrophils resulted in more patients in the immune cluster being classified as Non-responders.
Fig. 5
Pro-tumorigenic and anti-tumorigenic neutrophil signatures. A Feature plots depicting cells expressing Pro-tumor neutrophil (left) and Anti-tumor cluster 6 neutrophil (right) signatures in single-cell RNAseq data. B Comparison of Pro-tumor neutrophil (left) and Anti-tumor cluster 6 neutrophil (right) signatures between Responders and Non-responders in bulk RNA-seq data. C Survival curves in a large cohort (N = 631) for Pro-tumor neutrophil (left) and Anti-tumor cluster 6 neutrophil (right) signatures
留言 (0)