
記住我


In total, 114 articles were identified from the initial search, with 2 added after review. Following exclusion for duplicates or insufficient clinical details, 54 articles met the criteria and were deemed suitable for inclusion. The literature search identified 141 individual case reports of GS2 published in the literature until November 2023. In addition, 8 patients not previously described in the literature were identified from Great Ormond Street Hospital, London, United Kingdom for inclusion in the study following genetic diagnosis of RAB27A deficiency.
Demographic CharacteristicsIn total, 149 patients were included in the study. The median age at GS2 diagnosis was 1.5 years, with a wide range spanning from 10 days to 42 years. 74 were males (50%), 57 were females (38%), and data on sex were unavailable for 18 cases (12%). Patients of Qatari descent were most frequently found in our study cohort (n = 20, 14%), followed by Turkish descent (n = 15, 10%). Asymptomatic molecular diagnosis was made based on positive family history in 12 cases (8%). The demographic data of all patients is summarised in Table 1.
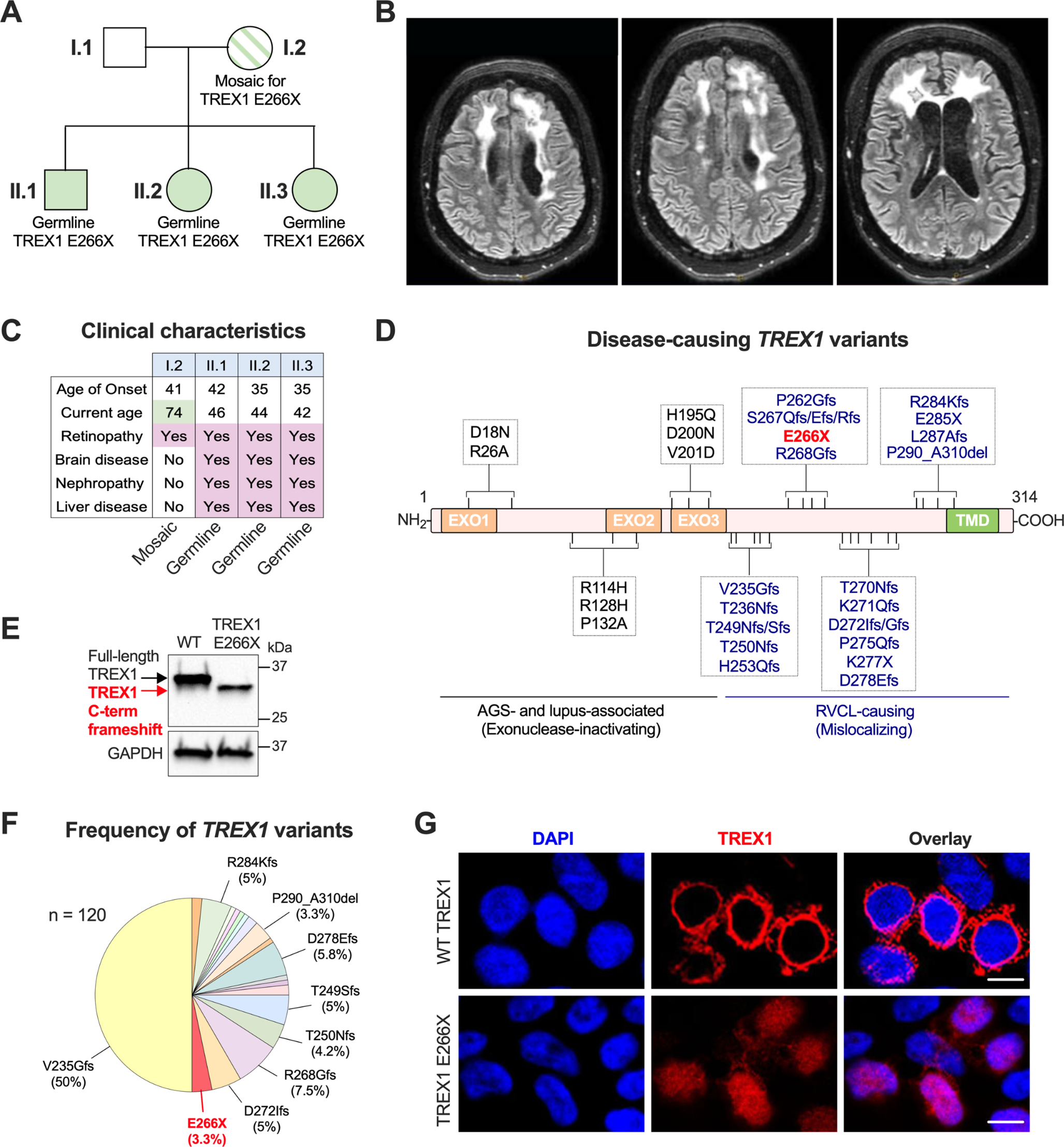
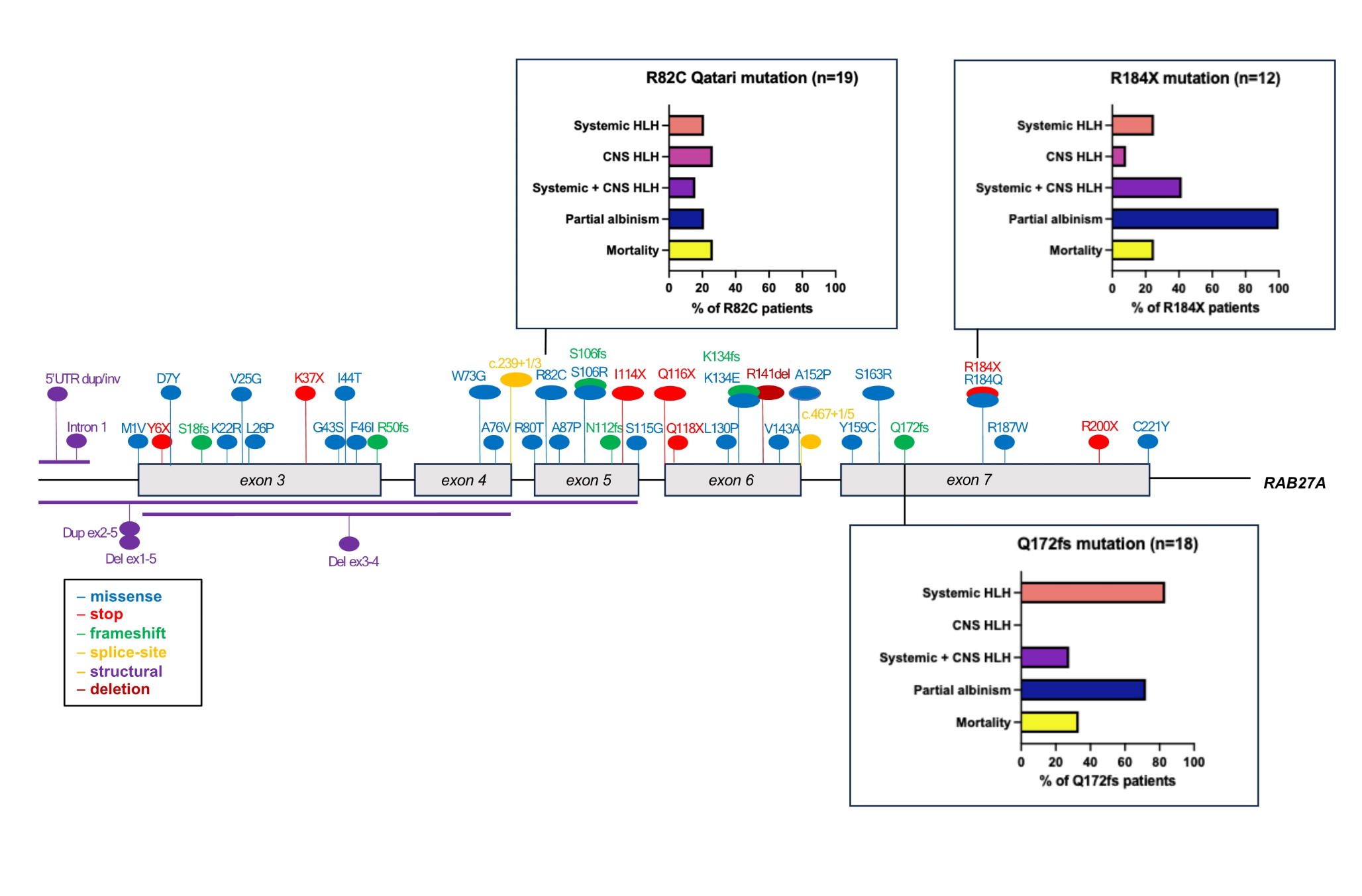
Table 1 Demographic data of patients with RAB27A deficiency. Number of samples is denominator where data were available. Median with interquartile range (IQR) shown in age of onset/death)Molecular AnalysisGenomic reports were available in 139 patients (93%). Overall, 56 distinct pathogenic sequence variants of RAB27A were identified, including intronic and structural variants. The mutations were distributed throughout the RAB27A gene, with several mutational hotspots identified (Fig. 1 and Supplemental Table 1). Analysis of haplotype blocks and literature review identified 3 potential founder mutations where genetic data were available: RAB27A c.244 C > T p.Arg82Cys, c.514_518del p.Gln172fs, c.550 C > T, p.Arg184X (NM_004580) [41, 71]. The RAB27A c.244 C > T p.Arg82Cys homozygous variant was only seen in Qatari patients. A shared haplotype block comprising 82 kilobases across the c.244 C > T p.Arg82Cys variant in 4 unrelated (Qatari) pedigrees. Shared haplotype blocks of 126 kilobases were found in 3 unrelated pedigrees across the c.514_518del p.Gln172fs variant and 5 unrelated patients with the c.550 C > T, p.Arg184X variant, suggestive of single recombination events from different common ancestors.
Fig. 1
RAB27A biallelic pathogenic variants. Founder mutations with phenotypic features depicted in bar charts. The first 2 exons of RAB27A are non-coding
Figure 1 shows the three founder mutations identified with different phenotypic profiles and outcomes. Parental consanguinity was reported in nearly two-thirds of the cases reviewed (n = 97, 65%) with biallelic mutations mostly homozygous, indicative of consanguinity and founder effects.
Clinical ManifestationsClinical features were described in all 149 patients reported. Presenting clinical features included fever (n = 94, 63%), splenomegaly (n = 92, 62%), hepatomegaly (n = 88, 59%), neurological features (n = 61, 41%) and muscle weakness or myalgia (n = 33, 22%), (Fig. 2A). Neurological features include signs such as ataxia, strabismus (n = 55, 37%), seizures (n = 21, 14%), and developmental delay or cognitive dysfunction (n = 15, 10%), A large proportion of patients had silvery-grey hair (n = 75, 50%) with a smaller number having hypopigmented skin (n = 32, 21%) (Fig. 2A). A minority of patients were asymptomatic (n = 12, 8%), having received molecular diagnosis following positive family history. A history of infection was noted in a quarter of the cases evaluated (n = 40, 27%), with more than half reporting frequent viral infections (Fig. 2A).
Fig. 2
A: Frequency of occurrence of clinical features. Values represent percentages of total cases (n = 149). B: Frequency of occurrence of diagnoses. Values represent percentages of total cases (n = 149)
Most patients developed HLH (n = 119, 80%). Information about HLH status was unavailable for 12 cases (8%) (Fig. 2B). Among the patients with HLH, nearly half presented with systemic HLH only (n = 52, 35%), combined systemic and central nervous system (CNS) HLH (n = 49, 33%), with the remaining cases presenting with isolated CNS HLH (n> = 18, 12%) HLH (Fig. 2B). HLH triggers were not adequately described in the literature to allow comment on this. Partial albinism was described in most patients as defined by the presence of one of the following: features of hypopigmented skin, silvery-grey hair, and uneven melanin pigmentation of the hair shaft detected by light microscopy (n = 105, 70%). 31 patients (21%) did not have any features of partial albinism. 5 patients in the cohort developed lymphoma which was successfully treated with chemotherapy (3%), with 2 specifically EBV-associated large B cell lymphoma [57, 58, 68].
Genotype-Phenotype CorrelationTo analyse genotype-phenotype correlation, the patients were divided into those with: (1) protein-truncating variants (PTV) (n = 56; 38%), expected to abolish Rab27a protein expression and function, and (2) hypomorphic variants (n = 41; 28%) such as compound heterozygous or biallelic missense mutations that are predicted to have residual Rab27a expression. Our analysis found patients with biallelic PTV were more likely to have HLH, particularly systemic HLH (24/56, 43%) and partial albinism (45/56, 80%), in comparison to hypomorphic variants (9/41, 22%; 20/41, 49%) (Fig. 3). However, isolated CNS HLH occurred more frequently in patients with hypomorphic variants (8/41, 20% compared to 2/56, 4%, p = 0.002). Only 1 patient with biallelic PTV did not have any features of partial albinism, compared to 19 patients with hypomorphic variants (2% compared to 46%, p = 0.001). The median age of onset of symptoms was 1.5 years (IQR = 0.3-5 years), and patients with PTVs presented at an earlier age (median age: 0.4 years, IQR = 0.25–2.7 years) than patients with biallelic hypomorphic mutations (median age: 5.4 years, IQR = 1.65-9 years) (X2 = 24.54, p = < 0.0001) (Fig. 4).
Fig. 3
Frequency of clinical features according to genotype groups. Compound variant types were excluded * denotes p-value < 0.05 from unpaired t-tests
Fig. 4
Onset of any symptoms in patients with PTVs and hypomorphic variants in Griscelli syndrome type 2
The most common pathogenic variant was the Qatari homozygous variant c.244 C > T, p.R82C which was identified in 19 of the study cohort (13%). This variant did not have typical features of GS2 as only 4 patients presented with systemic HLH (21%) and 4 patients had features of partial albinism (21%), while 5 had CNS HLH (26%). This contrasts with the founder variant c.514–518delCAAGC, p.Q172NfsX2 (n = 18) which had 15 patients presenting with systemic HLH (83%) and 13 patients with partial albinism (72%). Isolated CNS HLH was not found in any patients with the Q172fs founder variant and co-occurred with systemic HLH in 5 cases (28%) (Fig. 1). The Qatari variant R82C also differed from the c.550 C > T, p.R184X homozygous variant which is found in 12 patients in the study cohort. All 12 patients presented features of partial albinism, 3 developed systemic HLH (25%) and 5 CNS HLH (41%) (Fig. 1).
Characteristics of Late-Onset PresentationLate onset (at age ≥ 10 years) was noted in 16 cases (11%) (Fig. 5A) [13, 41]. 8 patients presented with homozygous missense hypomorphic variants (50%), while only 1 had a homozygous PTV (6%). The most common presenting feature in patients with late onset was systemic HLH (n = 10, 63%), with 8 patients also presenting with CNS HLH (50%) (Fig. 5B). Viral infections were found at HLH presentation in 8 patients (50%), with EBV implicated in 5 patients (31%). Late onset patients were more likely not to have partial albinism (n = 11, 69%) (Fig. 5B).
Fig. 5
(A) RAB27A pathogenic variants in patients who presented at 10 years and over. The first 2 exons are non-coding (B) Phenotypic characteristics of patients with Griscelli syndrome type 2 presenting at age 10 or older (n = 16)
Granule Release AssayGranule release assay results were available for 46 patients. 41 of 46 patients (88%) had reduced or absent granule release. In our local cohort (P1-8), we observed normal or equivocal granule release assay, and normal or equivocal NK cytotoxicity assay for P1-5 on repeated occasions, all patients being in an extended pedigree with Qatari R82C variant. Other R82C patients were found to have reduced NK cell cytotoxicity [11, 41].
Management ApproachFor HLH-directed therapy, 22 patients received HLH-94 protocol (22/119, 18%) whilst 15 received the HLH-2004 protocol (15/119, 13%) as first-line treatments. Data specific for CNS HLH treatment were limited in patients with GS2, but individual reports describe successful regimens have included the use of high-dose corticosteroids and mycophenolate mofetil (MMF) [56, 58].
HSCT was undertaken in less than one-third of published cases (n = 44, 26%). The most common donors for HSCT were matched unrelated donors (n = 17), followed by matched related, sibling donors (n = 5). Haploidentical/mismatched related donors were used in 4 patients (15%). 5 patients were transplanted following isolated CNS HLH (5/44, 11%) with 4 patients surviving. 5 patients were transplanted pre-emptively without a prior diagnosis of HLH (n = 5, 11%) (Table 2) [39, 41, 43].
Table 2 Characteristics of patients who had haematopoietic stem cell transplantation (HSCT). Subsections are given as percentage of number of patients where data is availableSurvival and Mortality OutcomesOverall, mortality was high (n = 50, 34%) in the study cohort. Data were not available to delineate specific causes of death or long-term outcomes in all patients. All patients who developed systemic HLH who did not receive HSCT shortly after, either died or were left with significant morbidity [43]. HSCT was undertaken in 44 patients with 18 having an uncomplicated post-HSCT course (41%) (Table 2). Donor information was only available in 25/44 patients (57%). Persistent neurological complications remained despite HSCT in 4 patients (9%), and fatal infections in the post-HSCT period occurred in 5 patients (11%). Among transplanted patients, 86% survived after HSCT (38/44). Of the 6 patients who died following HSCT, 5 had developed systemic and/or CNS HLH prior to HSCT (Fig. 6).
Fig. 6
Comparison of outcomes in patients who received HSCT to those who did not
留言 (0)