BRAF V600E mutations are present in tumors of histiocytic/dendritic cell origin with the highest frequency in LCH and ECD among histiocytic/dendritic cell neoplasms. The frequency of BRAF mutation in ECD in the lesion and peripheral blood monocytes has been reported to be between 51 and 67% [17, 19, 20]. Most large series recognize BRAF V600E mutation in 57% of LCH [21]. BRAF mutations have been reported in other histiocytic/dendritic cell neoplasms, but some may represent mixed histiocytosis. JXG has been reported to harbor BRAF mutation both in the CNS and ocular lesions [22,23,24]. Out of 10 pediatric CNS-JXG, 5 showed a BRAF mutation. However, after clinico-radiographic correlation, only 3 showed localized disease and others with systemic disease included pediatric ECD [24]. Most large studies report recurrent activating genetic alterations involving the classical MAPK signaling pathway in 57% of HS, but only a few reports either BRAFV600E or other BRAF mutations in HS [25, 26]. RDD has also been reported to show MAPK alterations in 30–50% of cases; however, no BRAF mutants were identified in RDD patients even in large studies [3, 27].
Recently, a few cases of RDD have been reported to harbor BRAF V600E mutations (Table 1). BRAF V600E has been reported in nodal RDD presenting with progressive bilateral cervical lymphadenopathy that improved without therapy [12]. A BRAF mutant RDD patient who presented with CNS involvement experienced treatment failure with steroids and died [13]. Recently, a patient with BRAF positive RDD who presented with CNS involvement responded to BRAF/MEK inhibitors [14]. RDD patients with BRAFY472C and BRAFR188G variants have been reported, but additional clinical features were not described [15]. A report of mixed RDD/LCH with BRAF mutation presented with diffuse lymphadenopathy, bone lesions, cerebellar lesions, and diagnostic RDD/LCH lesions in the lymph node and LCH lesion in the cerebellum [16]. Thus, to our knowledge, our case is only the second reported case of BRAF V660E mutated RDD associated with typical histologic and clinical findings.
Table 1 Reported BRAF-Mutated RDD or mixed histiocytosis with emperipolesisAlthough histiocytic/dendritic cell neoplasms are recognized by virtue of their histogenesis, some arise following the acquisition of somatic mutations in hematopoietic stem/progenitor cells (HSPCs). Xenotransplantation of CD34 + HSPCs from BRAF mutant patients with histiocytosis (ECD or MH) resulted in the development of histiocytosis-like lesions and detection of BRAF V600E in circulating monocytes of ECD and mixed histiocytosis patients. In addition, BRAF mutations were identified in CD34 + HSPCs in a subset of the same patients [29]. Studies of LCH patients identified BRAF mutations in HSPCs including common myeloid progenitors (CMP) and granulocyte–macrophage progenitors (GMP) [30]. Studies in LCH have shown BRAF mutations in the peripheral blood and CD34 + HSPCs of high-risk multisystem disease patients, while the presence of a BRAF mutation only in lesional tissue correlated with low-risk disease with a possible increased risk of recurrence [31, 32]. Given that HSPCs are the disease-initiating cells in ECD and LCH, it is thus not surprising that myeloid neoplasms develop in approximately 10% of LCH and ECD patients [33]. It will be interesting to evaluate both HSPCs and lesional cells in RDD patients to assess for the presence of BRAF mutations as well as correlate the pattern of BRAF mutation involvement with clinical outcomes, especially the development of myeloid neoplasms.
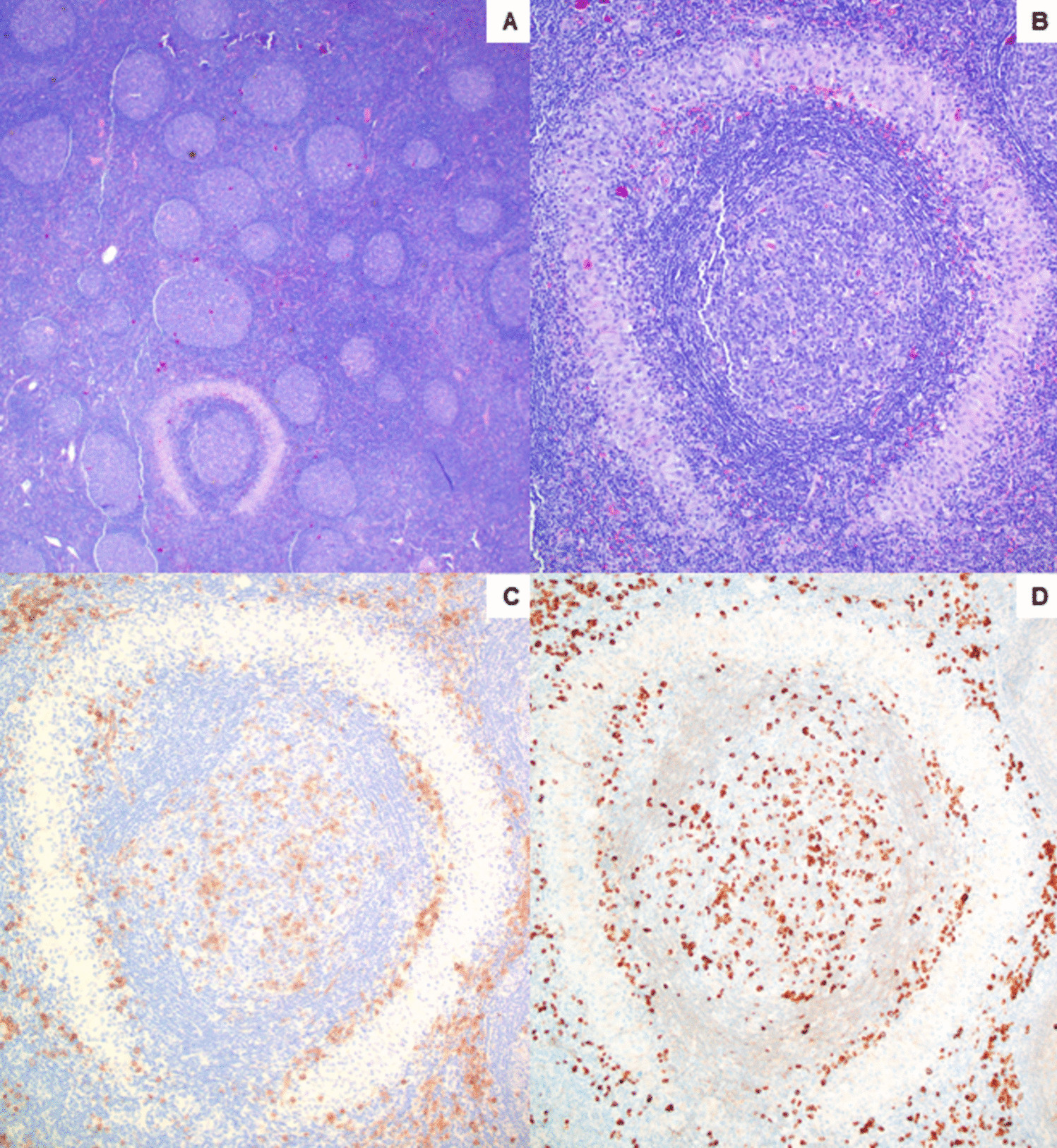
Given their overlapping morphologic and immunophenotypic features, the diagnostic approach to histiocytic/dendritic cell neoplasms should include a thorough review of clinical history. RDD usually presents as nodal disease but can be extranodal or multisystemic in 43% and 19% of cases, respectively. Cutaneous or CNS involvement can also be seen infrequently. Bone involvement occurs in 5–10% of RDD cases, typically in association with nodal disease, similar to this case. Bone lesions in RDD typically occur in the metaphysis or diaphysis and are osteolytic or mixed lytic/sclerotic [2]. Therefore, lesions in the femurs and tibia should raise concern for ECD since bone lesions in ECC are nearly always present as symmetric diaphyseal and metaphyseal osteosclerosis in the distal ends of the femurs and the proximal and distal tibia. Imaging studies are helpful, as ECD is frequently associated with dense infiltration of perinephric fat based (“hairy kidney”) as well as circumferential soft-tissue sheathing of the thoracic and abdominal aorta and its branches (“coated aorta”) [34]. Therefore, patients with similar histologic features presenting with single or multisystem lymphadenopathy most likely have RDD (similar to the patient in our case), while those with bone lesions and retroperitoneal or vascular findings without nodal involvement likely have ECD.
Cases of mixed histiocytosis that include both ECD and RDD components are difficult to ascertain since they both share similar histologic and immunophenotypic features, suggesting that at least some cases of RDD may have similar pathogenesis to ECD. Given the wide variety of presentations, it is unclear whether RDD represents a single disease. Emperipolesis can be associated with ECD and more frequently with MAP2K mutation. ECD with RDD-like lesions presenting as intra-abdominal and retroperitoneal lesions attributed to MAP2K1 mutation similar to three cohorts in which the frequency of RDD histology in ECD was reported as 3.6% (6/168), 3.1% (3/96), and 2.2%% (2/89) in overlap cases with typical clinical findings of bone lesions and/or retroperitoneal findings and/or coated vessels on imaging studies. In one cohort, 9 of 13 cases harbored MAP2K1 mutations, but none had BRAF mutations [10, 11]. Cases with CNS or cardiac involvement are particularly problematic. BRAF mutations have been described in a case characterized by CNS involvement with histologic evidence of emperipolesis recognized as ECD/RDD overlap disease [28], as well as in a case presenting with multiple bone lesions and pericardial effusion reported as ECD with emperipolesis [35].
RDD has an unpredictable clinical course and is fatal in 5–10% of cases, and thus, MAPK/RAF inhibitors have been used to treat RDD given the frequency of MEK pathway mutations. Cournoyer et al. treated 34 histiocytosis with favorable response in 94% of cases receiving either dabrafenib and/or trametinib after failure of chemotherapy and all cases receiving the inhibitor as first-line treatments. However, only 1 of 2 RDD patients with no identifiable mutation responded [36]. In a study of 16 RDD cases including KRAS- and MEK-variant cases, treatment with cobimetinib was associated with 88% vs. 38% overall response rates in mutant vs. wild-type RDD [37]. These studies led to FDA-approval of cobimetinib for the treatment of RDD and MEK inhibitors, which are essentially considered the first line of therapy for such patients [38]. Immunotherapy is usually not necessary in RDD cases since their clinical behavior is akin to a low-grade indolent histiocytic neoplasm. However, PD-L1 expression by cases of atypical cases of RDD exhibiting more aggressive behavior might justify treatment with immunotherapy, although this would require more clinical studies.
Given that vemurafenib induces responses in BRAF mutant ECD or LCH at high rates [39,40,41], it is not surprising that BRAF or MAPK inhibitor therapy has been described in BRAF mutant RDD. Cronin et al. treated one patient with a BRAF-mutated RDD CNS tumor [14], while Mastropolo et al. treated a case of BRAF V600E RDD and LCH with cytarabine, then dabrafenib. There was no response to either agent, and therefore, trametinib was added for concurrent MEK inhibition [16]. Diamond et al. found in their phase II trial that 18 patients with histiocytic neoplasms treated with cobimetinib had an 89% overall response rate; this trial included two patients with RDD, although neither harbored BRAF mutations [42].
The PD1/PD-L1 pathway regulates the balance between the stimulatory and inhibitory signals that regulate T cell responses needed for immune defense and tolerance. Ligation of PD-1 by PD-L1 activates a critical immune checkpoint leading to T cell dysfunction, exhaustion, and tolerance. High-affinity anti-PD-1 or anti-PD-L1 monoclonal antibodies (mAbs), which block their interactions, can reverse the immune checkpoint, releasing the brake on T cell responses [43]. PD-L1/PD-L2 can be expressed by normal or tumor-associated macrophages/ histiocytes. Neoplastic cells in histiocytic neoplasms also can express PD-L1, but checkpoint protein expression in RDD is variable, with studies reporting positivity in 2/11 (18%) [44], 0/4 [45], or 16/28 (57%) of cases [46]. It should be noted, however, that none of these studies evaluated BRAF mutant RDD. While immunotherapy would not be a typical therapeutic consideration in RDD since it typically behaves as low-grade indolent neoplasm and also responds to MEK inhibitors, it is intriguing to consider checkpoint blockade as a possible adjunct to MEK or BRAF inhibitor therapy in more severe cases of RDD.










留言 (0)