
記住我
Non-small-cell lung cancer (NSCLC) remains a major global health challenge due to its prevalence and high mortality rates regardless of the advancements in therapeutic approaches.[1,2] NSCLC spreads metastatically and substantially contributes to its poor prognosis and limited treatment options.[3] Epithelial–mesenchymal transition (EMT) represents a fundamental process where cancer cells acquire mesenchymal characteristics, which are implicated in tumor metastasis; this process promotes invasion, migration, and metastasis to distant organs.[4,5] The dysregulation of signaling pathways, including the epidermal growth factor receptor (EGFR) pathway, crucially contributes to the progression and metastasis of NSCLC.[6]
The EGFR signaling cascade exhibits frequent dysregulation in NSCLC, with genetic alterations, such as mutations and amplifications, that lead to constitutive activation of EGFR and downstream signaling pathways.[7,8] In addition to encouraging the growth and survival of tumor cells, this dysregulated EGFR signaling also aids in the production of EMT and the spread of metastatic disease.[9,10] However, the precise molecular mechanisms underlying EGFR-driven EMT and metastasis associated with NSCLC remain incompletely understood.
A member of the 5’-3’ exoribonuclease family, 5’-3’ exoribonuclease 2 (XRN2) is linked to the spread and metastasis of cancer.[11] XRN2 participates in RNA degradation and turnover processes, which influence gene expression and cellular behaviors.[12] Although XRN2 serves as a putative oncogene in several cancer types, including NSCLC,[13] its specific role in the regulation of EMT and metastasis mediated by EGFR lacks understanding.
This study is novel because it systematically investigates the role of XRN2 in NSCLC for the 1st time. Understanding the functional interplay between XRN2 and EGFR signaling in NSCLC pathogenesis can offer valuable insights into the molecular mechanisms underlying EMT and metastasis. Furthermore, elucidating the regulatory mechanisms governing XRN2 expression and activity in NSCLC may unveil novel therapeutic targets for treating metastatic diseases. Therefore, this study aimed to delineate the role of XRN2 in potentiating EMT and metastasis driven by EGFR in NSCLC. The findings provide a basis for developing targeted therapies to disrupt this axis and improve outcomes for advanced-stage NSCLC patients.
MATERIAL AND METHODS Animal experimentsThirty male BALB/c nude mice (6–8-weeks-old, 25 ± 2 g) were purchased from Bestest (Shenzhen, China). The housing and care of the animals adhered to legal requirements and national guidelines. The density of H460 cells was adjusted to 107 cells/mL, and 0.1 mL cell suspension was administered through tail vein injection. The mice were treated through transfection of short hairpin RNA-XRN2 (Sh-XRN2) or pCMV-XRN2 (Sangon Biotech, Shanghai, China) using lentivirus complex transfection solution. After 6 weeks, lung tissues from mice were collected and fixed in tissue fixative. The mice were euthanized through intraperitoneal injection of pentobarbital sodium (3 mg/mL) (110 mg/kg).
IHC stainingLung tissue specimens fixed in paraffin were sectioned into 4 mm-thick pieces. The tissue sections were dewaxed and rehydrated accordingly and heated in citrate buffer (70-PS0031, Multi Sciences, Hangzhou, China) at 100°C for 30 min to retrieve antigens. Utilizing 3% hydrogen peroxide, the tissue slices were treated to inhibit the endogenous peroxidase activity. Primary antibodies against Cluster of Differentiation 31 (CD31) (1:1,000 dilution, ab9498, Abcam, Cambridge, Germany) were treated with the tissue slices for an overnight period at 4°C. The tissue sections were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:1,000 dilution, ab6728, Abcam, Cambridge, Germany) for 1 h at room temperature in the dark. The sections were stained with 3,3’-diaminobenzidine (91-95-2, Sigma–Aldrich, St. Louis, Missouri, USA), counterstained with hematoxylin (517-28-2, Sigma–Aldrich, St. Louis, Missouri, USA), and sealed with Ramsan glue. Finally, images of the sections were examined and captured with a microscope (BX46, Olympus, Tokyo, Japan). The data were quantitatively analyzed by Image J (version 1.5f, National Institutes of Health, Maryland, USA).
Cell culturesHuman NSCLC cell line H460 (iCell-h160) and normal human bronchial epithelial cells BEAS-2B (iCell-h023) were obtained from iCell Biological Science Company (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle medium (iCell-0001, iCell Biological Science Company, Shanghai, China) and supplemented with 10% fetal bovine serum (FBS) (iCell-0500, iCell Biological Science Company, Shanghai, China) and 1% penicillin–streptomycin (iCell-15140-122, iCell Biological Science Company, Shanghai, China). The culture dishes were incubated in a cell culture incubator at 37°C with 5% carbon dioxide (CO2). After the cells reached a particular cell density, the culture media was changed every 2–3 days, and the cells were passaged. The cell lines used in this study have all undergone short tandem repeat authentication and tested negative for mycoplasma.
Cell transfectionRecombinant lentiviruses encoding specific shRNA against human XRN2, pCMV-vector, pCMV-XRN2, and pCMV-EGFR were designed and prepared by GeneChem (Shanghai, China). They included the following constructs: ShRNA negative control (Sh-NC) (sense: 5’-GATCCCCGTTCTCCGAACGT GTCACGTTTCAAGAGAACGTGACACGTTCG GAGAATTTTTGGAAA-3’, antisense: 5’-AGCTTT TCCAAA AATTCTCCGAACGTGTCACGTTCTCTTGAAACG TGACACGTTCGGAGAACGGG-3’), Sh-XRN2 (sense: 5’-GATCCCCGAAGCAGCAGTACTCGGAACTCGA GTTCCGAGTACTGCTGCTTCTTTTTGGAAA-3’, antisense: 5’-AGCTTTTCCAAAAAGAAGC AGCAGTACT CGGAAGCTCGAGTTCGAGTACTGCTGCTTCGG G-3’), pCMV-vector-blank, pCMV-XRN2 (sense: 5’-ATC GATGCTAGCGATCG TACGATCG-3’, antisense: 5’-CGA TCGTACGATCGCTAGCATCGAT-3’) and pCMV-EGFR (sense: 5’-ATCGATGCTAGCGATCGTACGATCG-3’, antisense: 5’-CGATCGTACGATCGCTAGCATCGAT-3’). The gene interference sequence was transfected into the cells using Lipofectamine 2,000 (11668019, Invitrogen, Carlsbad, California, USA).
Cell co-cultureHuman umbilical vein endothelial cells (HUVECs) and H460 cells, which have been successfully transfected, were seeded into separate wells of a culture plate. The cells were cultured until they reach 70% confluence. The HUVECs were placed in the lower compartment of a Transwell insert, and the H460 cells were placed in the upper compartment. The co-culture was incubated in a cell culture incubator (37°C with 5% CO2). Cell growth and interactions were regularly observed under a microscope (CX53, Olympus, Tokyo, Japan).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)Total RNA was isolated from the specimens utilizing the TRIzol extraction kit (DP424, TIANGEN, Beijing, China). Reverse transcription was performed by adding reverse-transcription reagents and RNA templates to the reaction mixture, which converted RNA into complementary DNA with the use of reverse transcriptase (R211-01, Nuoweizan, Nanjing, China). A reverse transcription polymerase chain reaction (RT-PCR) instrument (CFX96 Touch, Bio-Rad, Hercules, California, USA) was used to amplify the polymerase chain reaction (PCR) reaction mixture, with the fluorescence signal intensity recorded for each PCR cycle. RT-PCR data were subsequently analyzed using Bio-Rad CFX Manager (version 5.x, Bio-Rad, Hercules, California, USA). The relative expression levels of target genes were determined based on a threshold cycle of the fluorescence signal, which is typically in reference to a control gene. Analysis results of RT-PCR data were interpreted by comparing the expression levels of target genes under various conditions with those of the control group to draw conclusions from the experimental findings. The relative messenger RNA (mRNA) expression levels were calculated using the 2−ΔΔCt formula. The primer sequences in the study are shown in Table 1.
Table 1: Primer sequences employed in this investigation.
Primer name Prime sequences (5'-3') XRN2-F TGGATTAGGTTTACTGGCATCA XRN2-R GCAAGTACCCGTCCATCATAG VEGFA-F CTCAC ACACACACCAACCAGG VEGFA-R GAAGA AGCAGCCCATGACAG β-Actin-F CCTGGCACCCAGCACAAT β-Actin-R GGGCCGGACTCGTCATACT Western blotThe lysis buffer (R0010, Solarbio, Beijing, China) was mixed with the samples in order to break down cell membranes and liberate proteins. The test technique for bicinchoninic acid was utilized to quantify the lysed proteins. Electrophoresis was used to separate equal amounts of protein samples that were put onto sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels (P1200, Solarbio, Beijing, China). The isolated proteins were transferred from the gel to polyvinylidene difluoride membranes (YA1700, Solarbio, Beijing, China). The membranes were then placed in a blocking buffer, which typically consisted of 5% bovine serum albumin (SW3015, Solarbio, Beijing, China), to block non-specific binding. The blocked membranes were incubated with specific primary antibodies XRN2 (1:1,000 dilution, ab72181, Abcam, Cambridge, Germany), vascular endothelial growth factor A (VEGFA) (1:1,000 dilution, ab1316, Abcam, Cambridge, Germany), EGFR (1:1,000 dilution, ab52894, Abcam, Cambridge, Germany), phosphorylation (p)-EGFR (1:1,000 dilution, ab40815, Abcam, Cambridge, Germany), E-cadherin (1:1,000 dilution; cat no. A20798, ABclonal, Inc, Wuhan, China), N-cadherin (1:1,000 dilution; cat no. A19083, ABclonal, Inc, Wuhan, China), vimentin (1:1,000 dilution; cat no. A19607, ABclonal, Inc, Wuhan, China), and glyceraldehyde 3-phosphate dehydrogenase (1:1,000 dilution, ab8245, Abcam, Cambridge, Germany). These antibodies were bound to the target proteins. The membranes were washed with the wash buffer to remove primary antibodies that were non-specifically bound. Then, the membranes were incubated with HRP-conjugated secondary antibodies, which bound to the primary antibodies attached to the target proteins. The membranes were washed again with the wash buffer to remove secondary antibodies that were non-specifically bound (1:1,000 dilution; catalogue numbers: ZB-2305, ZB-230, and ZSGB-BIO; Beijing, China). The protein–antibody complexes were detected through chemiluminescence (SW2010, Solarbio, Beijing, China) techniques. The grayscale values of protein bands were analyzed using Image J software (version 1.5f, National Institutes of Health, Maryland, USA).
Hematoxylin and eosin (H&E) stainingTissue samples from lung metastatic lesions were collected and fixed using a fixative (P1110, Solarbio, Beijing, China) to preserve tissue structures. The fixed tissues were then washed in a series of alcohols for dehydration and water removal. The dehydrated tissues were embedded in paraffin wax to provide support during sectioning. The embedded tissues were sliced into thin sections using a microtome (RM2235, Leica Biosystems, Nussloch, Baden-Württemberg, Germany). Paraffin was removed from the slides through immersion in xylene (X8010, Solarbio, Beijing, China). Next, the sections were rehydrated through a series of alcohol washes and stained with H&E solution (G1120, Solarbio, Beijing, China). The slides were cleared through immersion in xylene to remove any remaining alcohol, and they were prepared for mounting. Finally, a coverslip was placed over the stained tissue sections using a mounting medium. A light microscope (CX53, Olympus, Tokyo, Japan) was utilized to examine the cellular morphology and tissue structure of the stained tissue sections.
Transwell assay Cell migration and invasionThe Transwell apparatus (3421, Corning, Corning, New York, USA) was prepared using a Transwell insert with an 8 mm pore size. For invasion, matrigel (G8061, Solarbio, Beijing, China) was added to the upper chamber and incubated at 37°C for 30 min to allow the Matrigel to solidify. To the lower chamber of a 24-well plate, 600 mL of media containing 10% FBS was introduced as a chemoattractant. The cells were suspended in serum-free medium, and the cell concentration was adjusted to (1 × 105 cells/mL). After the Matrigel had solidified, 100 mL of the cell suspension was added to the upper chamber. The apparatus was incubated at 37°C in a 5% CO2 incubator (Thermo Scientific Forma Series II, Thermo Fisher Scientific, Waltham, Massachusetts, USA) for 12 h. Following incubation, the non-invaded cells and Matrigel were gently removed from the upper chamber with a cotton swab. The invaded cells were fixed in the lower chamber with 4% paraformaldehyde (P1110, Solarbio, Beijing, China) for 10 min. Subsequently, the cells were stained with crystal violet (G1062, Solarbio, Beijing, China) for 10 min, and the excess stain was washed off with PBS. Finally, a microscope (CX53, Olympus, Tokyo, Japan) was used to examine and count the cells that had moved to the lower chamber. Cell migration and invasion results were quantitatively analyzed using Image J software (version 1.5f, National Institutes of Health, Maryland, USA).
Tube formation assayEach well of a 96-well plate was inoculated with an appropriate amount of Matrigel (M8370, Solarbio, Beijing, China) for complete coverage of the well bottom. The plate was incubated in a 37°C cell culture incubator until the matrix solidified. Thereafter, the treated cell suspension (1 × 105 cells/mL) was added to the pre-coated 96-well plate. The plate was incubated in a cell culture incubator (Thermo Scientific Forma Series II, Thermo Fisher Scientific, Waltham, Massachusetts, USA) at 37°C and 5% CO2 for specified 4–24 h to facilitate the formation of tube-like structures by the cells. Tube formation within the plate wells was observed using a microscope (CX53, Olympus, Tokyo, Japan). Tube formation was quantitatively analyzed using Image J software (version 1.5f, National Institutes of Health, Maryland, USA).
Statistical analysisStatistical analysis was performed utilizing GraphPad Prime software (version 8.0, GraphPad Software, San Diego, California, USA, https://www.graphpad-prism.cn/). Two datasets were compared using the t-test, and comparisons between numerous groups were made using the one-way analysis of variance. Subsequent post hoc analyses were conducted using Tukey’s method. The results were presented as mean ± standard deviation, and statistical significance was considered at P < 0.05.
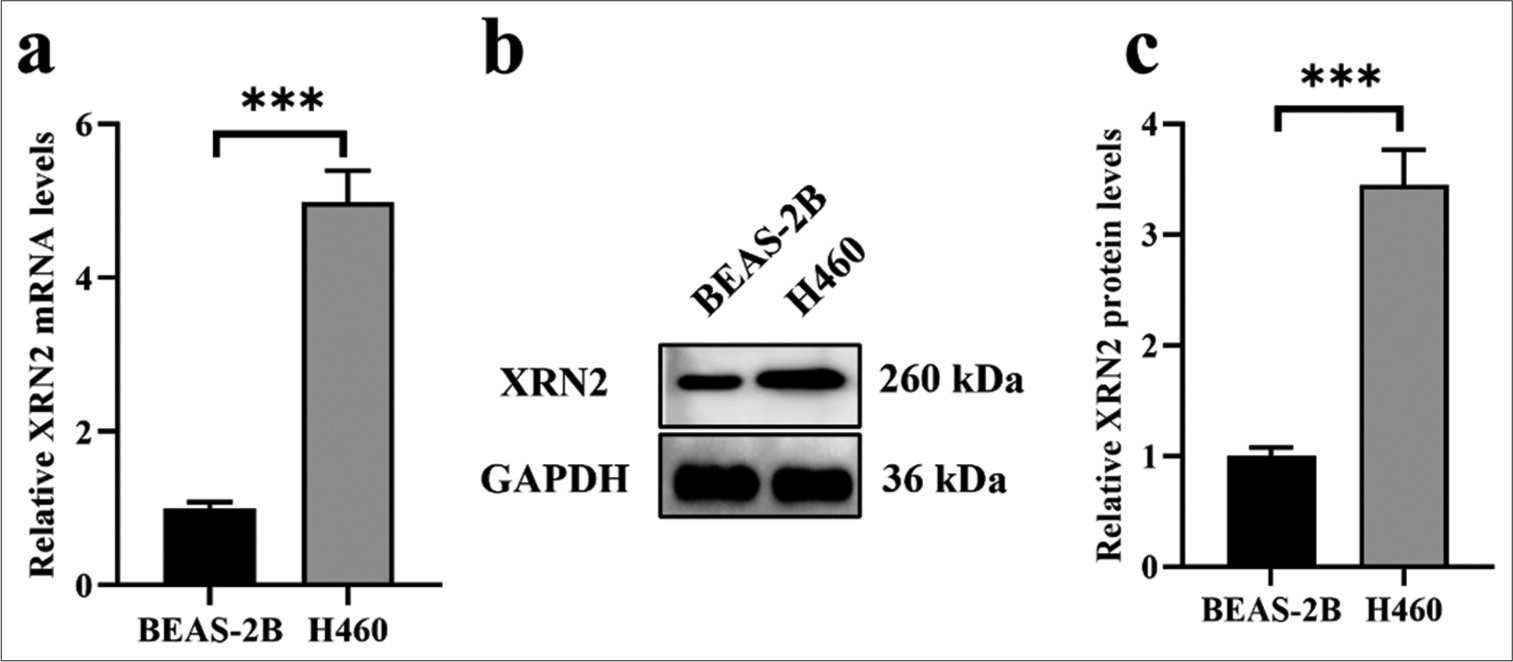
RESULTS XRN2 was upregulated in NSCLCWe assessed the expression of XRN2 in normal human bronchial epithelial cells (BEAS-2B) and an NSCLC cell line (H460) to investigate its expression profile. Our findings revealed the notable upregulation of XRN2 mRNA expression in H460 cells compared with that in the BEAS-2B cells (P < 0.001), [Figure 1a]. Similarly, XRN2 in H460 cells exhibited significantly elevated protein expression levels relative to that in BEAS-2B cells (P < 0.001), [Figure 1b and c].
Export to PPT
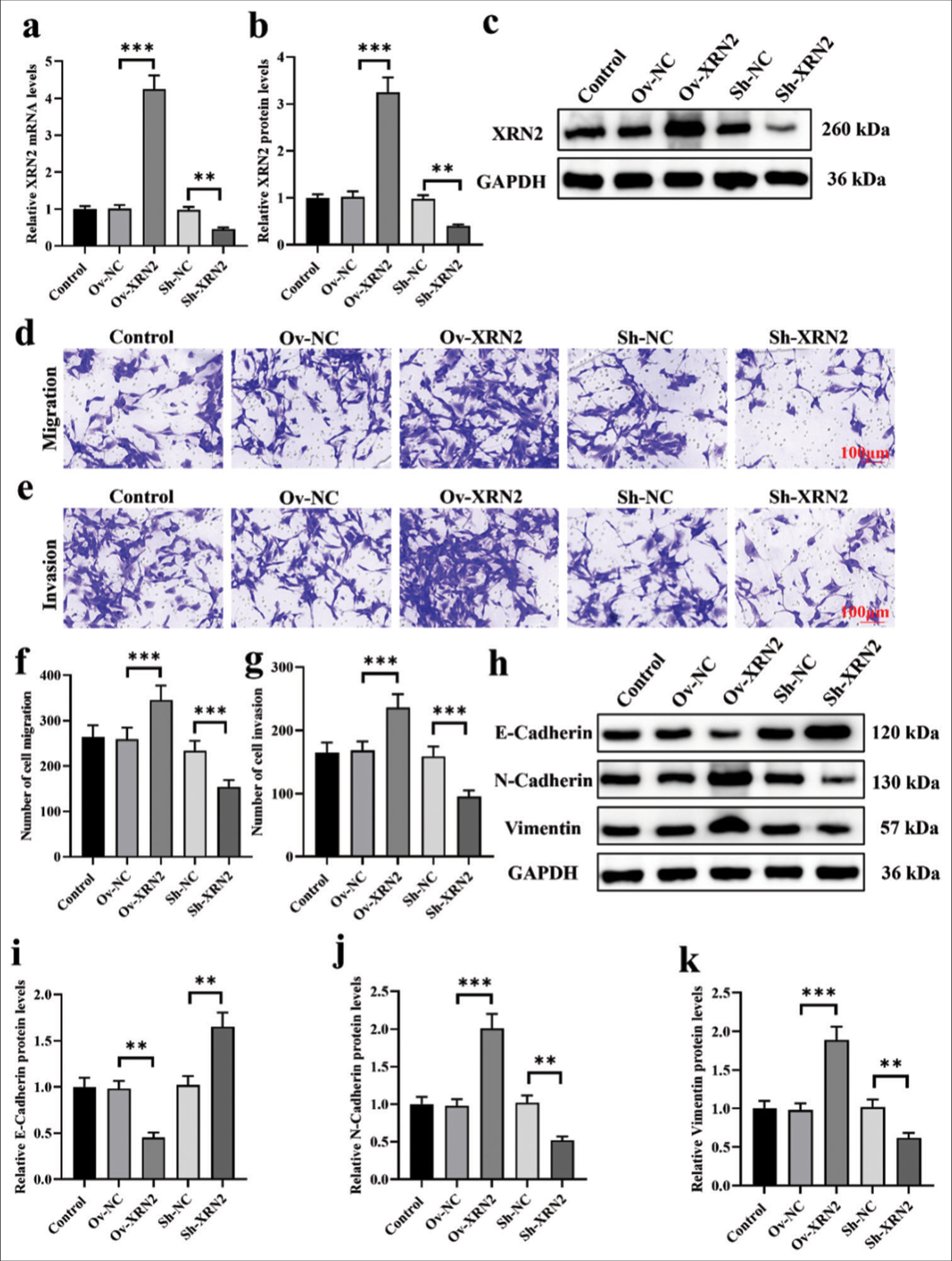
XRN2 promoted migration and the EMT process in NSCLC cellsWe established models of XRN2 overexpression and knockdown in H460 cells. [Figure 2a-c] confirmed the transfection efficiency of Ov-XRN2 and Sh-XRN2 (P < 0.001 and P < 0.01, respectively). Our findings indicate that XRN2 overexpression augmented the migratory and invasive capabilities of H460 cells, and XRN2 knockdown exerted the opposite effect (P < 0.001), [Figure 2d-g]. As observed, the overexpression of XRN2 markedly suppressed the protein expression of E-cadherin (P < 0.01) and substantially upregulated those of N-cadherin and vimentin (P < 0.001). Conversely, XRN2 knockdown significantly elevated the expression of E-cadherin protein and downregulated the expression levels of N-cadherin and vimentin (P < 0.01), [Figure 2h-k].
Export to PPT
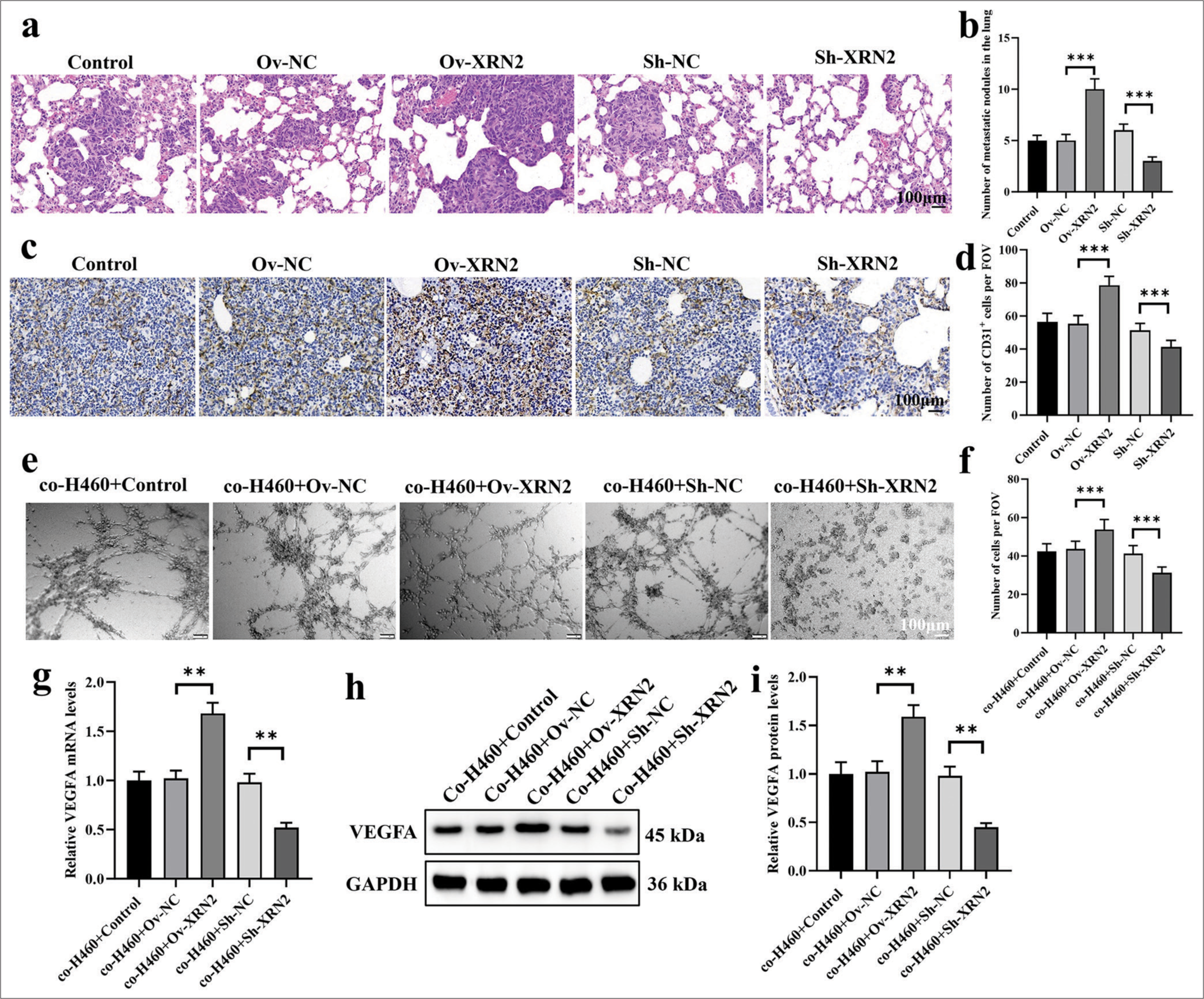
XRN2 promoted angiogenesis in the metastatic process of NSCLCWe examined the H&E-stained images of lung metastatic lesions in mice. Figure 3a shows that the XRN2 level significantly increased the number of lung metastatic lesions (P < 0.001) The XRN2 knock-down group showed a significantly reduced number of lung metastatic lesions (P < 0.001). Figure 3b results show that, the proliferation of tumor cells in the Ov-XRN2 group was significantly increased, the tissue arrangement was orderly, and the staining intensity was markedly enhanced. In the meantime, the proliferation in the Sh-XRN2 group was significantly reduced, the tissue arrangement was disordered, and the staining intensity decreased.
Export to PPT
Immunostaining was conducted for CD31 to characterize the aggregation of HUVECs in lung metastatic tumors. Figure 3c and d show that the XRN2 level significantly increased the proportion of CD31 + cells (P < 0.001), while the knockdown of XRN2 significantly decreased the proportion of these cells (P < 0.001). These findings imply that XRN2 overexpression improved the aggregation of HUVECs and facilitated angiogenesis in NSCLC lung metastasis.
We also explored the influence of co-H460 + XRN2 on the tube formation ability of HUVECs [Figure 3e and f]. Our findings indicate that HUVECs co-cultured with co-H460-Ov-XRN2 cells displayed considerably improved tube formation. Conversely, the HUVECs co-cultured with coH460-Sh-XRN2 cells exhibited significantly diminished tube formation ability. In addition, the relative expression levels of VEGFA in HUVECs support the observed phenomenon in tube formation assay. Specifically, the mRNA and protein expression levels of VEGFA in the co-H460-Ov-XRN2 group were markedly higher (P < 0.01) [Figure 3g-i]. Conversely, the mRNA and protein expression levels of VEGFA in HUVECs co-cultured with co-H460-Sh-XRN2 cells were significantly lower (P < 0.01), [Figure 3g-i]. In summary, these findings suggest that XRN2 indirectly promoted angiogenesis.
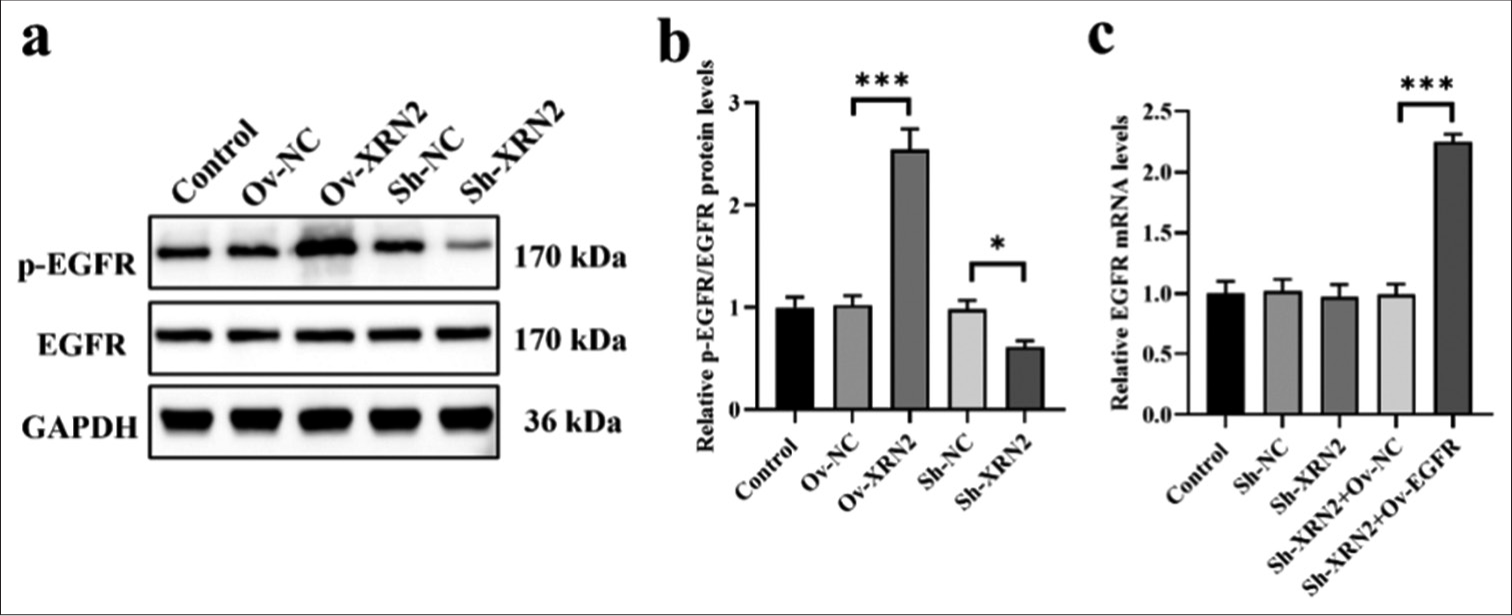
Overexpression of XRN2 promoted EGFR phosphorylation in NSCLC cellsXRN2 overexpression significantly enhanced the phosphorylation of EGFR in H460 cells (P < 0.001). Conversely, knockdown of XRN2 led to a significant decrease in the p-EGFR levels in H460 cells (P < 0.05), [Figure 4a and b]. However, the expression levels of EGFR remained unaffected by XRN2 knockdown and overexpression. These data suggest that XRN2 promoted the phosphorylation of EGFR. We co-transfected Sh-XRN2 and EGFR overexpression (Ov-EGFR) into H460 cells and validated the expression levels of EGFR mRNA through qRT-PCR analysis [Figure 4c] to further investigate the involvement of EGFR in the regulation of biological behaviors of H460 cells by XRN2. The results show no significant differences in EGFR mRNA levels among the Control, Sh-NC, Sh-XRN2, and Sh-XRN2+Ov-NC groups. However, the EGFR mRNA levels were significantly increased after EGFR overexpression treatment (P < 0.001), [Figure 4c].
Export to PPT
EGFR mediated the biological functions of XRN2 in NSCLC metastasisWe overexpressed EGFR to demonstrate the reversal of attenuated Sh-XRN2-induced biological functions in H460 cells. In addition, we explored the participation of EGFR in mediating the effects of XRN2 on NSCLC cell migration, invasion, and HUVEC tube formation. The H460 cells were co-transfected with Sh-XRN2 and OvEGFR. Our observations reveal that EGFR overexpression restored the inhibitory influence of XRN2 knockdown on NSCLC cell migration and invasion (P < 0.001), [Figure 5a-d]. Furthermore, Ov-EGFR downregulated the protein expression of E-cadherin (P < 0.001) and upregulated those of N-cadherin and vimentin in H460-Sh-XRN2 cells (P < 0.001), [Figure 5e-h]. The results of Figure 5i and j indicate that the tube formation ability of HUVECs in the co-H460+Sh-XRN2 group is significantly reduced (P < 0.001). The tube formation ability of HUVECs in the co-H460+Sh-XRN2+Ov-EGFR group was significantly higher (P < 0.001). These findings imply that EGFR mediated the functions of XRN2 in NSCLC.
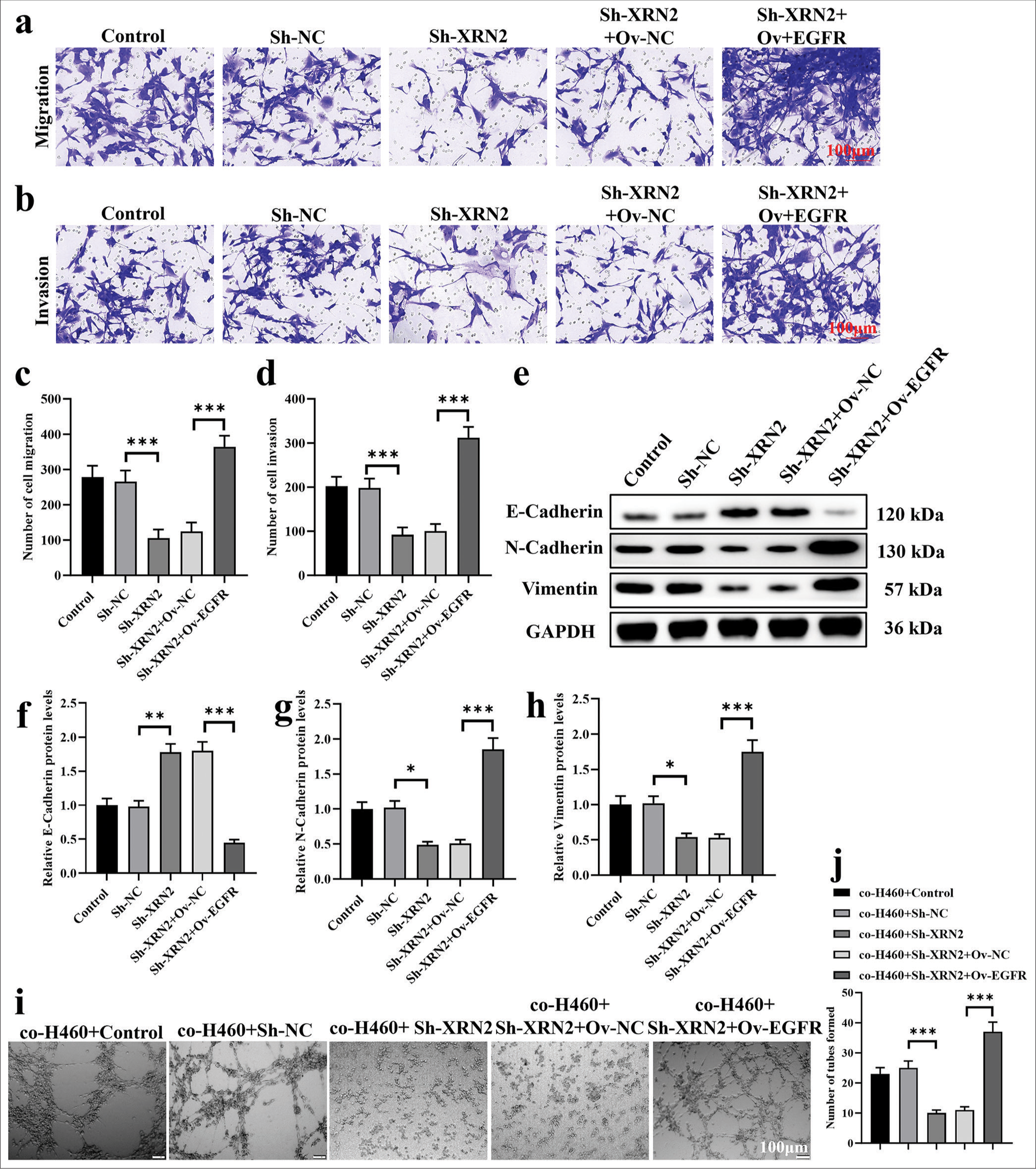
Export to PPT
DISCUSSIONCompared with those of previous studies, our research not only confirmed the expression pattern of XRN2 in NSCLC but also further elucidated its crucial role in the development and metastasis of NSCLC.[13] Notably, our study extends this understanding through systematically investigating the action mechanism of XRN2 in NSCLC and its effects on tumor cell migration, invasion, and angiogenesis for the 1st time.
Our study unveiled several remarkable findings. First, we not only observed the heightened expression of XRN2 in NSCLC cells but also delved into its role in the EGFR signaling pathway. Moreover, we explored its regulatory action toward EMT and tumor angiogenesis and offered novel perspectives on NSCLC pathogenesis. Furthermore, our investigation revealed that XRN2 overexpression significantly amplified EGFR phosphorylation, which fostered tumor cell migration, invasion, and angiogenesis. This discovery aligns with prior research indicating the effect of XRN2 on tumor cell behavior through EGFR pathway modulation. Prior research documented the irregular expression patterns of XRN2 across various cancer types and their close correlation with tumor invasiveness and metastasis.[14,15] Our study also concentrated on the interplay between XRN2 and EGFR and unveiled potential therapeutic targets. By leveraging in vitro and in vivo models, including assays for cell migration, invasion, and angiogenesis, we comprehensively evaluated the role and mechanisms of XRN2 in NSCLC. The findings not only deepen our understanding of XRN2 in NSCLC but also furnish vital theoretical and experimental ground works for future therapeutic strategies that target XRN2.
XRN2 exhibits an aberrant expression across various cancer types and a close association with tumor development and metastasis.[16] However, the different cancer types cause variation in the specific roles of XRN2.[17] Analyzing the expression levels, functions and regulatory mechanisms of XRN2 across various cancer types can provide valuable insights into its mechanisms of action and potential clinical applications for treating various cancer types.
XRN2 not only shows an association with the EGFR signaling pathway but also possibly interacts with other signaling pathways, such as Wnt.[18,19] These signaling pathways participate in tumorigenesis and development and collectively regulate the biological behaviors of tumor cells along with XRN2. Investigating the crosstalk between XRN2 and these signaling pathways can aid in the discovery of its broad mechanisms of action in tumor initiation and progression. Moreover, the tumor microenvironment substantially influences tumor progression, metastasis, and treatment resistance. Notably, XRN2 may participate in shaping the formation and function of this microenvironment.[20] In particular, XRN2 may influence the expression and activity of tumor-associated fibroblasts, immune cells, and endothelial cells, which affects the biological behavior of tumor cells.[11,21-23] Our research findings indicate XRN2 as a potential target for treating NSCLC. Further, prospective clinical and translational studies are needed to assess XRN2 as a potential clinical therapeutic target and provide theoretical and experimental evidence for its development based on personalized treatment strategies. Other studies indicated the possible association of XRN2 with drug resistance in tumor cells.[24] Exploring the mechanisms by which XRN2 contributes to the development of tumor cell drug resistance can offer new targets and strategies for overcoming such property.
In summary, our study underscores the pivotal role of XRN2 in NSCLC and provides novel theoretical and experimental foundations for developing targeted therapeutic strategies against XRN2. However, despite our crucial findings, additional clinical research and validation are needed to establish XRN2 as a potential therapeutic target in clinical practice.
SUMMARYThis study highlights XRN2 as a critical oncogene in NSCLC. The overexpression of XRN2 enhances tumor cell migration, invasion, and angiogenesis, which is possibly through modulating the EGFR signaling pathway.
AVAILABILITY OF DATA AND MATERIALSThe data that support the findings of this study are available from the corresponding author on reasonable request.
ABBREVIATIONSEMT – Epithelial–mesenchymal transition
NSCLC – Non-small-cell lung cancer
XRN2 – 5’-3’ exoribonuclease 2
EGFR – Epidermal growth factor receptor
CD31 – Cluster of differentiation 31
HUVECs – Human umbilical vein endothelial cells
HRP – Horseradish peroxidase
FBS – Fetal bovine serum
VEGFA – Vascular endothelial growth factor A.
AUTHOR CONTRIBUTIONSHZ and XCW: Designed the study, all authors conducted the study; MGW and YHC: Collected and analyzed the data; YHC and HZ: Participated in drafting the manuscript, and all authors contributed to critical revision of the manuscript for important intellectual content. All authors gave final approval of the version to be published. All authors participated fully in the work, take public responsibility for appropriate portions of the content, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or completeness of any part of the work are appropriately investigated and resolved.
留言 (0)