
記住我


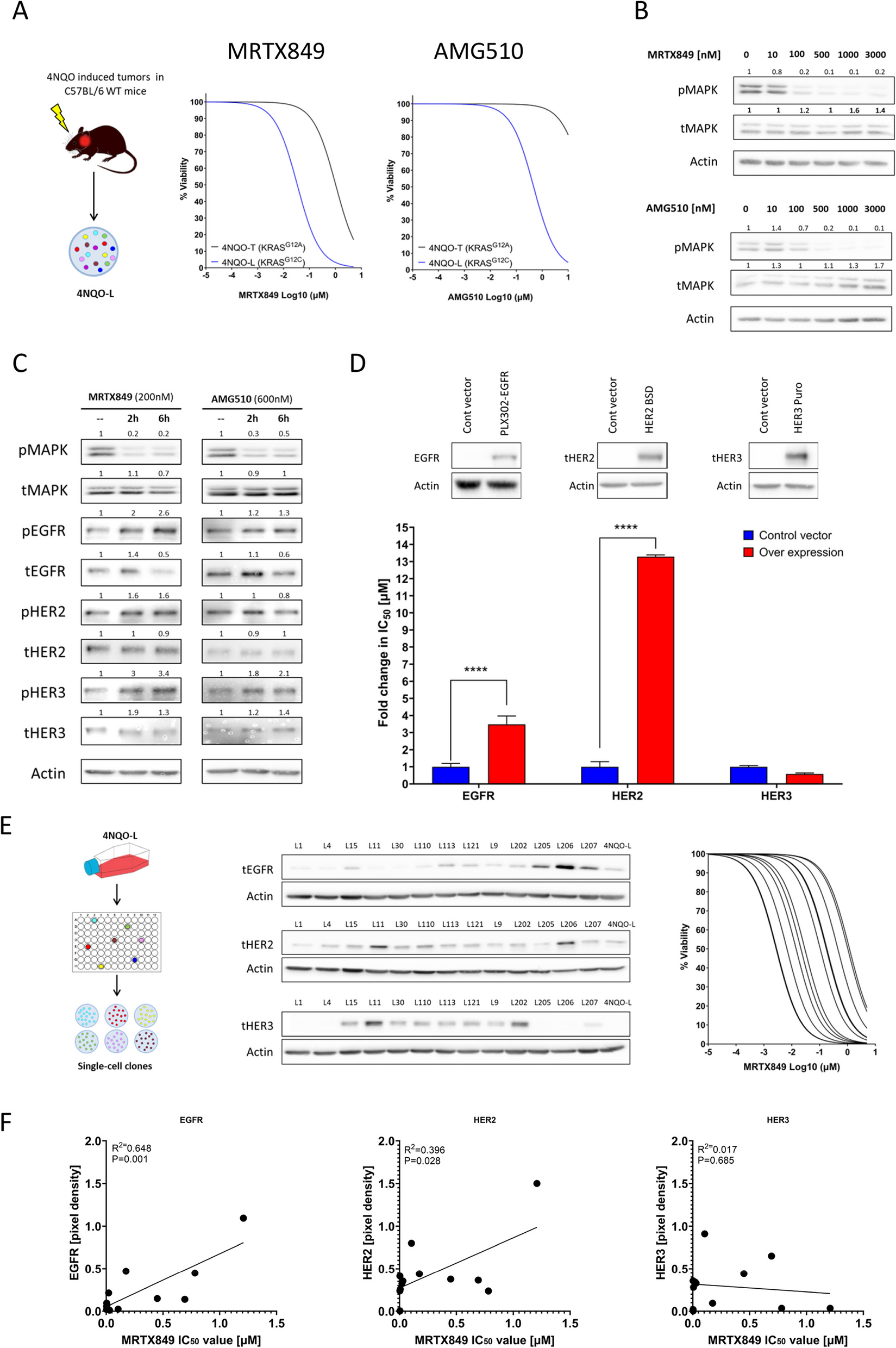



We accidently noticed MDM2 amplification by NGS testing in a panel of NSCLC patients who failed on Osimertinib treatment. Notably, MDM2 amplification was detected as a solely additional genetic alteration after Osimertinib progression, without other defined resistance mutations. Although EGFR activating mutations still existed in the repeated biopsy samples, these MDM2 amplified tumors failed to respond to Osimertinib (Fig. 1A). Patients with de novo MDM2 amplification also rapidly progressed on Osimertinib (Fig. 1B). Thus, we speculated that amplification of MDM2 may contribute to resistance to Osimertinib in EGFR mutant NSCLC.
Fig. 1
Identification of MDM2 amplification in NSCLC with resistance to Osimertinib. (A) The 61-years old female with stage IVB EGFR exon21 L858R mutant adenocarcinoma progressed on gefitinib due to T790M mutation. She began to receive Osimertinib as anti-cancer treatment in December 2016 and developed resistance in September 2019. The NGS testing of the repeated biopsy specimen showed the EGFR activating mutation and T790M mutation still existed after disease progression. However, MDM2 amplification was noted after Osimertinib resistance. (B) A 58-years old man with stage IVB lung adenocarcinoma referred to our institution in June 2023. The NGS result showed EGFR exon19 del and de novo MDM2 amplification. The patient was treated with first-line Osimertinib 80 mg per day and rapidly progressed on the treatment. Chest CT examination in September 2023 showed the emergence of multiple metastatic lesions indicated by red arrows. (C) Venn diagram showing the overlapping (purple part) of EGFR mutant NSCLC (red circle) and MDM2 amplified NSCLC (blue circle) according to NGS datasets consisting of 6,093 cases. (D) A patient cohort consisting of 241 cases of NSCLC concurrently harboring EGFR activating mutations and MDM2 amplification was analyzed. Tumor samples were arranged from left to right. Alterations of cooccurring genes were annotated for each sample according to the color panel below the image. The somatic mutation frequencies for each candidate gene were plotted on the right panel. Enrichment analysis and ranking of cooccurring genes in GO terms was performed using the online Metascape bioinformatic tool
To validate this hypothesis, we first screened the MDM2 state in a total number of 6,093 cases of NSCLC in our NGS datasets. Among these patients, 3,160 cases (51.86%) harbored an EGFR activating mutation (Fig. 1C). Aberrance of MDM2 was found in 375 cases (6.16%), in which the frequency of SNP and amplification was 0.53% and 5.63%, respectively. Thus, the most prominent aberration of MDM2 in NSCLC is genetic amplification. Of noted, 241 patients concurrently harbored EGFR mutation and MDM2 amplification (Table 1). These patients tended to display distinct clinicopathological features, such as no smoking history, adenocarcinoma histology, and more advanced disease. Genetic profiling suggested an enrichment of tumor promoting gene sets (including CDK4, RBM10 and GLI1) in the EGFRMut/MDM2Amp cohort (Fig. 1D), while these candidate genes did not directly affect cell proliferation or cell death programs, we therefore concluded that MDM2 may not regulate sensitivity to Osimertinib at transcriptional level.
Osimertinib resistant cells overexpressing MDM2We modeled acquired resistance to Osimertinib by generating polyclonal acquired resistant cell pools on the basis of stepwise dose escalation over a period of 10 days followed by maintenance in 1 µmol/L of Osimertinib over 3 months. We isolated 7 resistant clones and tested MDM2 expression individually and found clone #3 (denoted by PC-9 OR cells) yielded significantly elevated MDM2 transcripts when compared with parental PC-9 cells (Fig. S1). To characterize whether MDM2 is sufficient to drive resistance to Osimertinib, we overexpressed MDM2 in a panel of NSCLC cell lines harboring EGFR activating mutations. In comparison with NSCLC cells overexpressing empty vector as a negative control (NC), MDM2 readily induced a stable resistant phenotype to Osimertinib in PC-9 and HCC827 cells, as demonstrated by MTT cell viability assay and colony formation assay (Fig. 2A–C, Fig. S2A-S2C). Consistently, aberrant MDM2 expression also conferred resistance to Osimertinib in the T790M-positive H1975 cells (Fig. S2D-S2F). To explore whether MDM2 also promoted resistance to Osimertinib in vivo, we stably expressed MDM2 in PC-9 cells and seeded the cells in the flanks of nude mice. Treatment was initiated when the tumor nodules reached approximately 100 mm3. It was noted that PC-9 NC nodules were highly responsive to treatment, whereas the PC-9 MDM2 nodules continued to grow despite administration of Osimertinib (Fig. 2D). At the end of experiment, the average size and weight of xenograft tumor in PC-9 MDM2 group was 12-fold and 5-fold than that in PC-9 NC group, respectively (Fig. 2E F). Therefore, aberrant expression of MDM2 is sufficient to drive resistance to Osimertinib.
Fig. 2
Overexpression of MDM2 drives resistance to Osimertinib in EGFR mutant NSCLC. (A-C) The parental PC-9 sensitive cells were engineered to stably express ectopic MDM2 or empty vector as a negative control (NC) and tested for the sensitivity to Osimertinib by MTT assay and colony formation assay, respectively. ***P < 0.001. (D-F) PC-9 cells stably expressing NC or MDM2 were seeded in nude mice and treated with Osimertinib (5 mg/kg) or equal amount of Vehicle. The tumor volume was routinely monitored. At the end of the experiment, the xenograft tumors were carefully removed, weighted and photographed. **P < 0.01 (G) PC-9 cells stably expressing NC or MDM2 were treated with increasing concentrations of Osimertinib (0, 0.1, 0.5, 1 µmol/L) for 6 h. After indicated treatment, whole cell lysate (WCL) was prepared following standard protocol and analyzed for the phosphorylation status of EGFR, Akt and Erk by immunoblotting. GAPDH was used as an equal loading control. (H) Representative images of EdU labelling assay. PC-9 NC and PC-9 MDM2 cells were treated with 1 µmol/L Osimertinib or equal amount of DMSO for 24 h. Cell proliferation was evaluated by EdU staining. Scale bar = 200 μm. NS: not significant
The PI3K/Akt and MAPK/Erk signaling has been extensively described as major proliferative outputs of tyrosine kinases, thus, restoration of PI3K/Akt and MAPK/Erk signaling is a well-established mechanism underlying resistance to targeted therapeutics [19]. In order to clarify whether MDM2-induced Osimertinib resistance follows this dogma, we treated NSCLC cells with increasing concentrations of Osimertinib and determined the phosphorylation state of Akt and Erk. As shown in Fig. 2G and Fig. S2G-S2H, Osimertinib dose-dependently suppressed the phosphorylation of EGFR, along with efficient suppression of PI3K/Akt and MAPK/Erk signaling, in NSCLC cells infected with a NC lentivirus. Immunoblotting analysis of cellular extracts from MDM2 overexpressing cells showed that Osimertinib treatment also blocked EGFR phosphorylation, as well as Akt and Erk phosphorylation, to a comparable level. These findings implied that MDM2 may drive resistance to Osimertinib without restoring PI3K/Akt and MAPK/Erk signaling, and it was convincible to believe that overexpression of MDM2 minimally augmented cell proliferation. Indeed, the cell proliferation EdU fluorescence labeling assay showed that Osimertinib suppressed the EdU-positive signal in MDM2 overexpressing cells to a similar magnitude of that in NC cells (Fig. 2H, Fig. S2I-S2J). Thus, MDM2 aberration promoted resistance to Osimertinib independent of PI3K/Akt and MAPK/Erk machinery.
MDM2 arrests cell apoptosis through stabilization of MCL-1 proteinAside from enhanced cell proliferation, defects in apoptosis program also enable cancer cells to escape targeted therapy-induced cell death. On the basis of the fact that MDM2 overexpression did not affect cancer cell proliferation, we sought to determine the apoptotic state in Osimertinib resistant NSCLC cells. In response to Osimertinib treatment, the sensitive cells underwent pronounced apoptosis, as measured by TUNEL assay, whereas the proportion of TUNEL-positive signal in PC-9 OR and other MDM2 overexpressing resistant cells was largely decreased (Fig. 3A, Fig. S2K-S2M). Because p53 participates in most biological events driven by MDM2, we therefore investigated whether the arrest in apoptosis was affected intracellular p53 content. We found that depletion of endogenous p53 in PC-9 OR cells minimally affected the sensitivity to Osimertinib (Fig. 3B–E). When the sensitive PC-9 cells were engineered to express a MDM2 truncated mutant that lacked the p53 binding domain (MDM2 ΔPBD), the resultant cells were still resistant to Osimertinib-induced apoptosis (Fig. 3G and I). As such, aberrant apoptotic response may contribute to Osimertinib resistance driven by MDM2, which was not likely caused by the canonical MDM2-p53 protein regulatory loop.
Fig. 3
MDM2 drives resistance to Osimertinib independent of the canonical MDM2-p53 regulatory loop. (A) Representative fluorescent images of TUNEL assay. PC-9 NC and PC-9 MDM2 cells were treated with 1 µmol/L Osimertinib or equal amount of DMSO for 24 h. Cell apoptosis was visualized and calculated by TUNEL assay. NS: not significant. Scale bar = 200 μm. ***P < 0.001. (B-E) The Osimertinib resistant PC-9 OR cells were transfected with siRNA targeting p53 to deplete endogenous p53 expression and evaluated for the sensitivity to Osimertinib by MTT assay, colony formation assay, and TUNEL assay, respectively. Scale bar = 200 μm. NS: not significant. (F-I) The Osimertinib sensitive PC-9 cells were engineered to stably express the MDM2 ΔPBD mutant that was unable to interact with p53. The resultant cells were treated with Osimertinib or DMSO. Therapeutic response to Osimertinib was determined by cytotoxic assays, including MTT, colony formation and TUNEL assay. Scale bar = 200 μm. NS: not significant. **P < 0.01. ***P < 0.001
To clarify this aberrant apoptotic response, we probed cell lysates with antibodies targeting BCL-2 family members known to facilitate apoptosis. Immunoblotting assay showed that Osimertinib treatment dose-dependently triggered a decline in anti-apoptotic proteins MCL-1, BCL-2 and BCL-xL in sensitive cells, together with increased expression of pro-apoptotic protein BAX, BIM and the fragmentation of caspase-3 and PARP. In PC-9 OR and other MDM2 overexpressing resistant cells, it was noted that Osimertinib suppressed BCL-2 and BCL-xL, and increased BAX and BIM protein, to a similar magnitude compared with sensitive cells. Notably, the MCL-1 anti-apoptotic protein was refractory to Osimertinib treatment and there was no detectable reduction in MCL-1 protein abundance following treatment (Fig. 4A, Fig. S2N, Fig. S3A-S3B). Because prompt MCL-1 protein destruction was crucial for apoptosis induction [20], we speculated that MCL-1 stabilization in these MDM2 overexpressing cells mediated resistance to Osimertinib.
Fig. 4
MDM2 arrests Osimertinib-induced cancer cell apoptosis through manipulating MCL-1. (A) PC-9 NC and PC-9 MDM2 cells were treated with increasing concentrations of Osimertinib (0, 0.1, 0.5, 1 µmol/L) for 48 h. Cell apoptosis indicators (PARP and Cleaved caspase-3) and BCL-2 family proteins involved in cell apoptosis program (MCL-1, BCL-2, BCL-xL, BAX, BIM) were analyzed by immunoblotting. GAPDH was used as an equal loading control. (B) PC-9 cells were treated with DMSO or 1 µmol/L Osimertinib for 48 h. To block proteasome-mediated protein degradation, MG132 at a final concentration of 20 µmol/L was added into the cell culture medium 8 h before cell harvest. MCL-1 protein level was determined by Western blot. (C) PC-9 MDM2 cells and PC-9 NC cells were split into 6 cm cell culture dishes and treated with 25 µg/mL CHX for indicated time intervals. Proteins of interest were separated by electrophoresis, transferred to nitrocellulose membrane and probed with indicated antibodies. (D) PC-9 OR cells were transfected with siRNA targeting MDM2 or NC. The half-life of candidate proteins was assessed by CHX chase assay. (E) Representative images of colony formation assay in PC-9 OR cells treated with DMSO, Osimertinib, AZD5991 or their combination. (F) Western blot analysis of PARP and caspase-3 fragmentation after 48 h of indicated treatment. (G) Evaluation of PC-9 OR cell apoptosis by TUNEL assay. The magnitude of apoptosis was calculated by the percentage of TUNEL-positive cells. Scale bar = 200 μm. (H-I) Cytotoxicity assessment of PC-9 MDM2 cells treated with DMSO, Osimertinib, AZD5991 or their combination. Scale bar = 200 μm. (J) Transient expression of the degradation resistant MCL-1 S159A mutant and its effect on cancer cell apoptosis induction in PC-9 OR cells treated with the Osimertinib + AZD5991 combinational strategy. **P < 0.01. ***P < 0.001
In support of this idea, we found Osimertinib primarily repressed MCL-1 protein through the ubiquitin-proteasome system, which could be restored by a proteasome inhibitor MG132 (Fig. 4B). The cycloheximide (CHX) chase experiments revealed that MCL-1 protein in sensitive cells was rapidly destabilized upon Osimertinib treatment, and its abundance drop to half of baseline within 2 h after stalling protein synthesis. Importantly, MDM2 antagonized Osimertinib-induced MCL-1 protein destruction and extended its half-life to different degrees (Fig. 4C, Fig. S3C-S3D), whereas depletion of endogenous MDM2 in PC-9 OR cells accelerated the degradation of MCL-1 protein (Fig. 4D). These results indicated MDM2 was required for the stabilization of MCL-1. To assess whether the stabilization of MCL-1 protein was sufficient to confer Osimertinib resistance, we treated PC-9 OR and PC-9 MDM2 cells with a combination of AZD5991, a selective and potent MCL-1 inhibitor [21]. While most Osimertinib or AZD5991 single agent-treated resistant cells remained alive and formed cell colonies, a significant percentage of cells underwent apoptosis upon their combination (Fig. 4E and I, Fig. S3E-S3F). In contrast, transient expression of a degradation resistant MCL-1 S159A mutant in PC-9 OR cells arrested cell apoptosis even in the presence of combinational treatment (Fig. 4J). These data implied that MDM2 arrested Osimertinib-induced cancer cell apoptosis at the step of defected MCL-1 destruction.
Overcoming osimertinib resistance by dual inhibition of EGFR and MDM2Although inhibiting MCL-1 could overcome resistance to Osimertinib in our experimental setting, the clinical application of MCL-1 inhibitors is challenging due to undesirable hematologic and cardiac toxicity [22]. We therefore sought to answer whether concurrently targeting EGFR and MDM2 as an alternative strategy to overcome Osimertinib resistance. We knockdown endogenous MDM2 in PC-9 OR cells with siRNAs oligos and found Osimertinib perturbed cell viability and led to a noticeably reduction in colony number (Fig. 5A). Results from TUNEL assay also yielded cancer cells responded to Osimertinib and underwent apoptosis after MDM2 depletion (Fig. 5B). Immunoblotting analysis showed the reversal of resistance was accompanied by MCL-1 destabilization (Fig. 5C), suggesting that MDM2 launched a defense mechanism against MCL-1 destruction in response to Osimertinib.
Fig. 5
Overcoming resistance to Osimertinib by targeting MDM2. (A-B) The PC-9 OR cells were seeded in 6 well plate and transfected with siRNA targeting MDM2 or NC. Representative images of colony formation assay and TUNEL assay after DMSO or Osimertinib treatment were shown. Scale bar = 200 μm. (C) The PC-9 OR cells transfected with MDM2 siRNA or NC siRNA were treated with increasing concentrations of Osimertinib (0, 0.1, 0.5, 1 µmol/L) for 48 h. Expression of indicated BCL-2 family members and fragmentation of PARP and caspase-3 were analyzed by immunoblotting. GAPDH was used as an equal loading control. (D-E) The PC-9 OR cells were seeded in 6 well plate and treated with DMSO, Osimertinib, Osimertinib + Nutlin-3, and Osimertinib + MX69 for 10 days. Representative images of colony formation assay and TUNEL assay after treatment were shown. Scale bar = 200 μm. (F) The PC-9 OR cells were treated with Osimertinib (0, 0.1, 0.5, 1 µmol/L) and MDM2 inhibitors (5 µmol/L Nutlin-3 or 5 µmol/L MX69) for 48 h. The apoptosis state and MCL-1 protein level were assessed by immunoblotting
Small molecule inhibitors targeting MDM2 expression, such as MX69 (Fig. S4A), and inhibitors targeting MDM2-p53 interaction, such as Nutlin-3, have been developed. To evaluated the feasibility of pharmacologically targeting MDM2 to overcome resistance, we treated PC-9 OR and PC-9 MDM2 resistant cells with a combination of MDM2 inhibitors. Intriguingly, MDM2-conferred resistance was only reversed by MX69 (Fig. 5D and E, Fig. S4B-S4C), which dissected MDM2 and invigorated MCL-1 destruction to boost an apoptotic response to Osimertinib. While Nutlin-3 mechanistically bound to the hydrophobic pocket domain of MDM2 to selectively disrupt its interaction with p53, leaving MCL-1 protein intact and functional (Fig. 5F, Fig. S4D), failed to reverse resistance. These features heightened the necessity of rational MDM2 inhibitors selection and strategy design for effective combinations.
The MDM2 E3 ubiquitin ligase and osimertinib resistance converge at FBW7The C-terminal RING domain possesses E3 ubiquitin ligase activity and is required for the proteolytic efficacy of MDM2. In our study, overexpression of MDM2 antagonized MCL-1 protein turnover, which seemed to be in sharp contrast to its pro-proteolytic activity. To gain insight into the biological event elucidating how MDM2 engaged MCL-1 protein stabilization, we generated the MDM2 ΔRING truncated mutant and the C464A mutant within the C-terminal RING domain that abolished its E3 ubiquitin ligase activity. Ectopic expression of these constructs defective in E3 ligase activity failed to induce a resistant phenotype, as judged by a panel of cytotoxicity assays (Fig. 6A and B). Immunoblotting analysis further demonstrated that cells underwent substantial apoptosis, together with accelerated MCL-1 proteolysis, when MDM2 was replaced by its E3 ligase activity deficient mutants (Fig. 6C). Thus, the E3 ubiquitin ligase activity of MDM2 was required for maintaining an Osimertinib resistant phenotype through stabilization of MCL-1 protein.
Fig. 6
MDM2 as a potential E3 ubiquitin ligase for FBW7 tumor suppressor. (A-B) The parental PC-9 cells were engineered to stably express the MDM2 ΔRING or MDM2 C464A mutants that lacked E3 ubiquitin ligase activity. Cells were seeded in 6 well plate and treated with DMSO or Osimertinib. Representative images of colony formation assay and TUNEL assay were shown. Scale bar = 200 μm. (C) PC-9 cells stably expressing MDM2 ΔRING or MDM2 C464A mutants were treated with increasing concentrations of Osimertinib (0, 0.1, 0.5, 1 µmol/L) for 48 h. Expression of indicated BCL-2 family members and fragmentation of PARP and caspase-3 were analyzed by immunoblotting. GAPDH was used as an equal loading control. (D) Sequence alignment of MDM2 recognition motif across FOXO3, HEXIM1, IRS1, HIPK1 and FBW7. The conservation of Q-enriched domain in the N-terminal proportion of FBW7 was assessed across different species. (E) The WCL of PC-9 cells was precipitated with anti-MDM2/anti-FBW7 antibody or equal amount of isotype IgG. The resultant immunoprecipitates were subjected to immunoblotting. (F) HEK293 cells were transfected with HA-FBW7 together with Flag-MDM2 construct. Forty-eight hours after transfection, WCL was prepared and precipitated with anti-HA/anti-Flag antibody or equal amount of isotype IgG. The cell lysates and immunoprecipitates were subjected to immunoblotting. β-actin was used as an equal loading control. (G) The effect of MDM2 on the ubiquitination status of FBW7. MDM2 was stably expressed in PC-9 cells. The ubiquitination level of FBW7 was determined by immunoprecipitation with an anti-FBW7 antibody and probed with an anti-Ub antibody. (H) HA-FBW7 and His-Ub, together with Flag MDM2 or its ΔRING and C464A mutant were transiently expressed in HEK293 cells. HA-FBW7 protein was pull down by an anti-HA antibody and probed with an anti-His antibody. (I) Myc-FBW7 and Flag-MDM2, together with His-Ub or its K48-only and K63-only construct were transfected into HEK293 cells. Forty-eight hours after transfection, Myc-FBW7 protein was precipitated with an anti-Myc antibody and probed with an anti-His antibody. (J) Flag-MDM2, His-Ub and HA-FBW7 or its ΔQ mutant were expressed in HEK293 cells. The ubiquitination status of HA-FBW7 was determined by immunoprecipitation. (K-L) The PC-9 OR cells stably expressing FBW7 or its ΔQ mutant were treated with increasing concentrations of Osimertinib and evaluated for cytotoxicity by MTT assay and apoptosis immunoblotting assay, respectively. *P < 0.05. **P < 0.01
We speculated that proteins complexed and ubiquitinated by MDM2 may be responsible for MCL-1 proteolysis. This was motivated by previous reports describing MCL-1 protein as a substrate of FBW7 E3 ubiquitin ligase, by which FBW7 directly ubiquitinates MCL-1 protein for degradation [20, 23]. It was therefore proposed that MDM2 E3 ubiquitin ligase may stabilize MCL-1 protein through manipulating FBW7 E3 ubiquitin ligase. To explore whether the interaction of MDM2 and FBW7 constituted the mechanistic basis for MDM2-mediated MCL-1 stabilization and Osimertinib resistance in NSCLC, we screened in silico prediction and literature reports and noticed a consensus recognition motif enriched in glutamine (Q) across a panel of MDM2 substrates. We grouped FBW7 amino acid sequence and found the Q-enriched motif within the N-terminal region was highly conserved among the orthologues in different vertebrate species (Fig. 6D), raising a possibility that MDM2 may restore MCL-1 protein abundance through binding and catalyzing FBW7 destruction. In agreement with this idea, we detected co-precipitation of endogenous MDM2 when FBW7 was captured by an antibody for IP. When reciprocal experiment was performed, FBW7 was pulled down and complexed with MDM2 (Fig. 6E, Fig. S5A-S5B). Likewise, Flag-tagged MDM2 and HA-tagged FBW7 were found to complex with each other upon transient expression in HEK293 cells (Fig. 6F). The binding to MDM2 resulted in K48-linked polyubiquitination, but not K63-linked polyubiquitination, of FBW7 protein (Fig. 6G and I, Fig. S5C-S5D), whereas FBW7 engineered to lose the Q-enriched consensus motif (ΔQ) failed to complex with MDM2 as efficiently as FBW7 (Fig. 6J). To this end, it is conceivable that ectopic expression of the ubiquitination resistant FBW7 ΔQ mutant in PC-9 OR resistant cells ensured prompt MCL-1 proteolysis and rescued sensitivity to Osimertinib (Fig. 6K and L, Fig. S5E).
Biological interaction between MDM2 and FBW7 and its clinical relevanceConsistent with a role as E3 ubiquitin ligase, overexpression of MDM2 accelerated the turnover of FBW7 protein in multiple NSCLC cell lines, along with the stabilization of proteins targeted by FBW7, such as MCL-1, cyclin E and c-Myc (Fig. 4C). In contrast, depletion of endogenous MDM2 enhanced FBW7 stability and prompted degradation of MCL-1, cyclin E and c-Myc (Fig. 4D), supporting the physiological role of MDM2 in regulating FBW7 protein abundance and biological functions. To precisely map the MDM2 ubiquitination site on FBW7, we used computational prediction that yielded the evolutionarily conserved Lys412 residue within C-terminal WD40 domain as a putative ubiquitination site targeted by MDM2. To validate this site of interest, we generated the FBW7 K412R mutant for ubiquitination analysis. Notably, the ubiquitination state of FBW7 was largely compromised when the Lysine was replaced by Arginine (Fig. 7A), supporting the Lys412 residue is indeed a major site for MDM2-mediated ubiquitination of FBW7.
Fig. 7
Targeting MDM2 is a feasible approach to overcome resistance to Osimertinib in vivo and its clinical relevance during Osimertinib progression. (A) HA-FBW7 or its K412R mutant was expressed in HEK293 cells. The ubiquitination status of FBW7 was determined by immunoprecipitation assay. (B-D) Representative images of PC-9 OR cells xenograft tumors grew in nude mice receiving Vehicle, Osimertinib (5 mg/kg), MX69 (20 mg/kg) and their combination. The tumor volume was routinely monitored. At the end of the experiment, the xenograft tumors were carefully removed, weighted and photographed. *P < 0.05, **P < 0.01, ***P < 0.001. (E) Evaluation of cell apoptosis in xenograft tumors by TUNEL assay. Scale bar = 200 μm. ***P < 0.001. (F) IHC staining of MDM2, FBW7, MCL-1 and cleaved caspase-3 in xenograft tumors. The enlarged window indicated protein intensity and distribution pattern. Scale bar = 100 μm. (G) Analysis of MCL-1 IHC score after indicated treatment. NS: not significant. **P < 0.01, ***P < 0.001. (H-I) A total number of 23 cases of surgical resected NSCLC were analyzed for MDM2 and FBW7 expression by IHC staining. Scale bar = 100 μm. Each NSCLC sample yielded an IHC score for MDM2 and FBW7, respectively. The correlation between MDM2 and FBW7 was determined by linear regression analysis. (J-K) IHC analysis of MDM2 expression in matched treatment-naïve and Osimertinib resistant NSCLC patients. Scale bar = 100 μm. NS: not significant. ***P < 0.001
Finally, we investigated the therapeutic efficacy of co-targeting MDM2 to overcome resistance to Osimertinib in vivo. PC-9 OR cells were subcutaneously seeded in nude mice and treated with Osimertinib, MX69, or their combination. It was noted that Osimertinib elicited very limited single agent anti-tumor efficacy to Osimertinib since the xenograft tumor continued to grow in the presence of Osimertinib and the tumor volume and tumor weight in the Osimertinib group was comparable to that in Vehicle group. Treatment with MX69 revealed modest anti-tumor activity with a slight decrease in tumor size and a slight increase in tumor weight. However, a significant reduction in xenograft tumor outgrowth was noticed when these two drugs were combined with each other (Fig. 7B–D). This combinational strategy was the most effective in reducing tumor volume and weight, probably mainly through the induction of extensive apoptotic cell death (Fig. 7E). Histological evaluation of the isolated xenograft tumor showed the drug combination strategy provoked the strongest inhibitory potency on MCL-1 protein expression and the strongest capacity on apoptosis induction, together with the breakdown of MDM2 oncoprotein and the restoration of FBW7 tumor suppressor (Fig. 7F and G). Indeed, the reverse correlation between MDM2 and FBW7 was clinically relevant. We evaluated the expression of MDM2 and FBW7 in 23 cases of surgical resected NSCLC by IHC staining and found that the MDM2low tumors tended to have a relative higher expression level of FBW7, whereas FBW7 expression in the MDM2high tumors was largely diminished (Fig. 7H). The regression analysis of corresponding IHC scores confirmed a negative correlation between MDM2 and FBW7 with a R value of -0.52 (Fig. 7I). Hence, MDM2 is a physical negative regulator of FBW7 in NSCLC. Finally, MDM2 overexpression as a mechanism of resistance to Osimertinib has been validated in repeated biopsy samples upon disease progression. Evaluation of MDM2 expression in paired treatment-naïve and treatment-failure specimens yielded higher MDM2 IHC scores after Osimertinib resistance (Fig. 7J and K). Collectively, these findings demonstrated that FBW7 expression in NSCLC is tightly regulated by MDM2 E3 ligase. The protein interaction between MDM2 and FBW7 definitely exists in clinical setting and their balance determines the sensitivity to Osimertinib in EGFR mutant NSCLC. Disruption of the balance, such as MDM2 amplification, promotes FBW7 protein destruction and impairs the cell apoptosis program to lead resistance to targeted therapy (Fig. 8). Thus, manipulating MDM2-FBW7 interaction would be a feasible approach to overcome resistance to Osimertinib in EGFR mutant NSCLC.
Fig. 8
A schematic diagram of MDM2-mediated resistance machinery and its implications for overcoming resistance to Osimertinib in EGFR mutant NSCLC. In EGFR mutant/MDM2low NSCLC, Osimertinib treatment triggers MCL-1 anti-apoptotic protein ubiquitination catalyzed by FBW7 E3 ligase that leads to MCL-1 protein destruction and substantial apoptosis. These MDM2low tumors manifest as Osimertinib sensitive NSCLC. However, when cancer cells gain high expression of MDM2, MDM2 acts as an E3 ubiquitin ligase for FBW7 to degrade this tumor suppressor, which results in the accumulation of MCL-1 anti-apoptotic protein. MCL-1 in MDM2high tumors is refractory to FBW7-mediated protein destruction and therefore cancer cells are resistant to Osimertinib-induced apoptotic cell death. Hence, targeting MDM2 to augment prompt destruction of MCL-1 protein would be a promising strategy to overcome resistance to Osimertinib in EGFR mutant NSCLC
留言 (0)