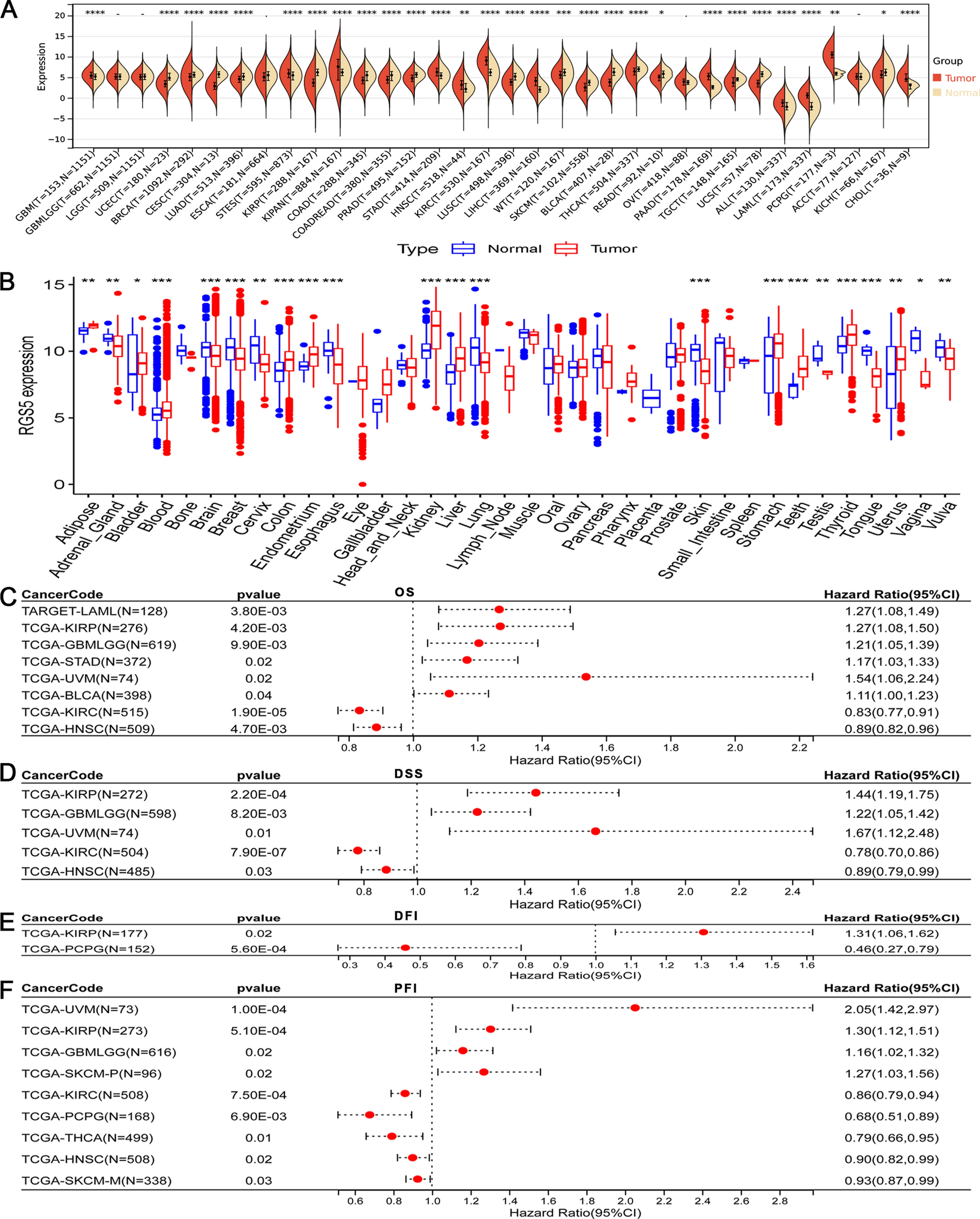
Despite the advances in reperfusion strategies and pharmacological treatments, AMI remains a persistent global health challenge [28]. Its rapid progression often results in critical delays in treatment, leading to severe and potentially fatal outcomes. In this context, the interplay between ferroptosis and immune dysregulation in particularly complex. Ferroptosis has been demonstrated to stimulate the proliferation and activation of immune cells such as B cells, T cells, and macrophages, which play a crucial role in the immune response to AMI [29]. For example, ferroptosis initiates the migration of neutrophils to the myocardium following heart transplantation, thereby contributing to cardiac injury in mouse models [12]. Glutathione peroxidase 4 (GPX4), a key regulator in the ferroptosis pathway, maintains cellular redox balance and significantly impacts immune responses associated with this form of cell death [30]. The release of Damage-Associated Molecular Patterns subsequent to ferroptosis can further influence the differentiation and function of immune cells in the context of tissue damage [31]. This underscores the importance of understanding and targeting ferroptosis in cardiovascular health. Gaining insights into GPX4's function within immune cells could reveal key determinants that drive ferroptosis in these cells. In this study, we identified 446 differentially expressed genes (DEGs) closely associated with immune disorders in AMI patients by analyzing the GSE60993 and GSE29532 datasets through GO and KEGG pathway analyses.
Further exploration of immune function alterations and immune cell abundance in AMI led to the identification of 11 core overlapping genes (DUSP1, TLR4, FTH1, SLC40A1, DDIT4, JDP2, LAMP2, NFS1, SLC1A5, PRDX1, and XBP) through a Venn diagram analysis of the 446 DEGs and 259 ferroptosis-related genes. These genes are significantly associated with immune cell infiltration, including CD8+ T cells, B cells, macrophages, and neutrophils, as demonstrated by correlation analysis. Additionally, these genes are linked to the inhibition of specific immune-related signaling pathways in AMI, such as interactions among lymphoid and non-lymphoid cells, primary immunodeficiency, and the adaptive immune system. Recent studies have further substantiated the connection between ferroptosis and immune cells like B cells, CD8+ T cells, macrophages, and neutrophils, lending further support to our findings [12, 31,32,33].
Furthermore, the suppression of cardiac ferroptosis by ferritin H in cardiomyocytes was recently highlighted as crucial in heart disease progression and failure [34]. Based on this, we evaluated the predictive potential of these 11 key genes for long-term heart failure in AMI patients. JDP2, DUSP1, and DDIT4 showed excellent diagnostic ability, with AUCs of 0.917, 0.875, and 0.736, respectively. Among them, only JDP2, DUSP1, TLR4, NFS1, and SLC1A5 were consistently identified as potential diagnostic markers across independent datasets, clinical samples, and in vitro cellular experiments.
Previous research has emphasized the essential role of macrophages in cardiac remodeling. An imbalance between pro-inflammatory M1 and anti-inflammatory M2 macrophage phenotypes can amplify inflammation, leading to increased cardiac injury [35]. Studies have also shown a complex interaction between macrophage polarization and ferroptosis at the cellular level [31]. Our findings indicate that macrophages are positively correlated with genes such as JDP2, TLR4, and DUSP1 but negatively correlated with SLC1A5 and NFS1. We hypothesize that these identified genes could be critical mediators of the interaction between macrophage polarization and ferroptosis, influencing the pathological progression of AMI.
Among these genes, DUSP1, which belongs to the autophagy modulator family, is primarily involved in MAPKs within cancer cells [36]. Elevated DUSP1 expression is associated with the suppression of autophagy-driven ferroptosis in pancreatic cancer [36]. Recent studies have also highlighted the role of cardiac DUSP1 in reducing infarct size and improving myocardial function through the JNK pathway [37]. Additionally, the nuclear receptor coactivator 4 (NCOA4) has been found to mediate chondrocyte ferroptosis via the JNK-JUN signaling pathway [38]. However, the potential involvement of DUSP1-mediated ferroptosis through the JNK pathway in AMI progression remains unexplored.
JDP2, a transcription factor within the leucine zipper superfamily, regulates both transcriptional repression and activation [39]. Animal studies have shown that increased JDP2 expression can exacerbate contractile dysfunction, induce atrial dilation and hypertrophy, and accelerate heart failure progression [40,41,42]. Our validation dataset supports these findings, with JDP2 demonstrating a strong diagnostic value (AUC = 0.917) for heart failure post-AMI [43]. While studies using genetically modified mice have conclusively demonstrated JDP2's role in heart failure and atrial fibrillation, human studies are still lacking [39]. Further exploration of JDP2's clinical implications in heart failure prediction is necessary.
DDIT4, alternatively referred to as DNA damage response 1 or stress-triggered protein, suppresses mTOR signaling by bolstering the tuberous sclerosis complex, a mechanism observed in several tumors including breast cancer, glioma, and gastric cancer [44,45,46,47]. Moreover, DDIT4 induction by IL-10 suppresses mTOR signaling while maintaining mitochondrial integrity [48]. A recent study unveiled that DDIT4 promotes endothelin-1 and hypoxia-inducible factor-1α, contributing to injury and fibrosis in systemic lupus erythematosus end-organs [49]. However, the role of DDIT4 in the context of hypoxia-inducible factor-1α/mTOR pathways and their impact on long-term heart failure post-AMI remains poorly understood.
TLR4, a part of the transmembrane receptor family, is prominently found on the membranes of macrophages, dendritic cells, endothelial cells, and epithelial cells [50]. TLR4 activation triggers pro-inflammatory responses, exacerbating inflammation and cardiac fibrosis [51]. Extracellular vesicles derived from pro-inflammatory macrophages have been shown to worsen cardiac dysfunction through the TLR4/NF-κB pathway [52]. TLR4/Trif-centered signaling also drives inflammatory responses post-cardiac transplantation, primarily through ferroptosis and cell death mechanisms [12]. Our findings align with contemporary research linking TLR4 to AMI [53].
NFS1 facilitates the biosynthesis of iron-sulfur clusters (Fe-S) by acting as a sulfur donor [54]. These clusters are essential cofactors for Fe-S proteins, which are involved in iron homeostasis, lipid synthesis, and energy metabolism [55]. NFS1 has also been shown to correlate significantly with immune cell infiltration levels [56]. However, its role in AMI has yet to be fully elucidated.
SLC1A5, often designated as ASCT2, belongs to the solute carrier family 1 and acts as a cell-surface solute-carrying transporter that regulates glutamine uptake [11]. Emerging research on the intracellular glutamine pool has highlighted their importance in protein translation, cell growth, mTORC1 signaling activation, and apoptosis prevention [57]. While SLC1A5 amplifies ferroptosis, its function in immune cells linked to AMI remains largely unclear.
Our study highlighted six relatively unexplored ferroptosis-associated genes—JDP2, DUSP1, DDIT4, TLR4, NFS1, and SLC1A5—with potential relevance to AMI. Among these, the identified associations of NFS1 and SLC1A5 with AMI represent novel findings in this field. A comprehensive understanding of these genes is crucial to confirm their relevance to AMI and their potential as therapeutic targets. Despite the significant findings of this study, several limitations must be acknowledged. First, the bioinformatics approach relies on the quality of public datasets, which may introduce biases. These findings must be rigorously validated through prospective, large-scale trials and functional studies in vitro and in vivo. Second, the use of the AC16 cell line as a model for ischemic conditions, although widely used, may not fully replicate the complex physiological environment of the human cardiac tissue. Additional in vivo studies or primary cardiomyocytes from human tissues would help confirm the generalizability of our findings. Third, our focus on ferroptosis-related genes addresses only one aspect of AMI pathogenesis; future research should consider other regulated cell death mechanisms, such as necroptosis and pyroptosis. These limitations should be taken into account when interpreting the results, and future work is needed to address these gaps.









留言 (0)