
記住我



Complex karyotype (CK), occurring in 10–15% of adult acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) cases, is associated with poor prognosis and frequently co-occurs with TP53 mutation [1,2,3]. TP53 mutations correlate with dismal outcomes regardless of the type of therapy received [3,4,5], underscoring the need to understand the biological background of the CK group. The genetic complexity of AML/MDS with CK complicates the detection of specific genetic alterations that drive cancer growth during tumorigenesis. Although transcriptomic studies have revealed the dysregulation of cell cycle-related pathways [6, 7], whether these signatures are specific to the CK group or are primarily because of TP53 mutations remains unclear.
Single-cell RNA-seq and the bulk gene expression deconvolution analysis has revealed that the presence of primitive and differentiated cell states in AML and MDS is highly variable, and that cell type composition may be associated with genetic features and therapy resistance in a cell type-specific manner [8,9,10,11]. A comprehensive analysis of CK cases using gene expression profiles, including the maturation states of leukemia stem and progenitor cells (LSPCs), is yet to be performed.
We hypothesized that the distinct prognostic features associated with TP53 mutations in AML/MDS with increased blasts (MDS-IB) patients with CK correspond to a unique genetic background. To test this, we integrated two public and one in-house bulk RNA-sequencing datasets to compare gene expression signatures and cellular hierarchy composition states of the CK groups according to TP53 mutation status. We correlated these genetic features with ex-vivo drug sensitivity results to identify potential differences in treatment response.
A total of 61 bone marrow (BM) aspirate samples were collected from patients with AML or MDS-IB (AML, 49 samples; MDS-IB, 12 samples) from our cohort (herein the KUMC cohort). This study was approved by the Institutional Review Board of Korea University Guro Hospital (2021GR0247, 2021GR0572). Informed consent was obtained in accordance institutional review board and exempt for data obtained retrospectively. The study was conducted in accordance with the Declaration of Helsinki and all methods were performed in accordance with the relevant guidelines and regulations. In addition, we included publicly available gene expression data from clinical AML datasets, including 289 AML samples from the BeatAML cohort [10] and 203 AML samples from the Leucegene cohort [6] (Figure S1). CK was defined as ≥3 unrelated chromosome abnormalities, excluding other class-defining recurrent balanced abnormalities [1,2,3]. In total, 155 samples with CK were collected from all cohorts (KUMC cohort: 26 samples; Leucegene cohort: 68 samples; BeatAML cohort: 61 samples).
Gene set enrichment analysis was performed, and cellular hierarchy composition was also analyzed to classify samples into four cell types (Primitive, granulocytic–monocytic progenitor (GMP), Mature, and Intermediate) [11]. Patient-derived ex-vivo drug sensitivity results from the BeatAML cohort were analyzed using the area under curve (AUC) metric, where a low AUC indicated sensitivity and a high AUC indicated resistance. Detailed methods are provided in the supplementary material.
Of the 533 AML/MDS-IB samples, 155 (29.0%) were identified with CK. As expected, TP53 was the most frequently mutated gene, detected in 64.5% (n = 100) of the CK samples, followed by NRAS (14.2%), NF1 (10.3%), TET2 (9.7%), RUNX1 (9.7%), and DNMT3A (9.7%) (Supplementary Fig. S2A, B). While the prevalence of RAS pathway-related gene mutations did not differ significantly between TP53mut CK and TP53wt CK (P = 0.569) groups, myelodysplasia-related genes mutations [12] were significantly more prevalent in TP53wt CK compared to TP53mut CK (76.4% vs. 25.0%; P < 0.001), indicating the possibility of MDS background (Supplementary Fig. S2C, S2D).
We analyzed the outcomes of patients with CK using clinical data from the KUMC and BeatAML cohorts. The baseline characteristics of the CK and non-CK groups are shown in Supplementary Tables 1 and 2. CK was associated with poor outcomes in patients with AML (BeatAML cohort, P < 0.001), as well as in patients with AML/MDS-IB (KUMC cohort, P = 0.005) (Supplementary Fig. S3A). Within the CK group, the presence of TP53 mutation was associated with reduced survival in the BeatAML cohort (P = 0.001) (Supplementary Fig. S3B). TP53 mutation did not significantly affect outcomes in the AML/MDS-IB group in the KUMC cohort.
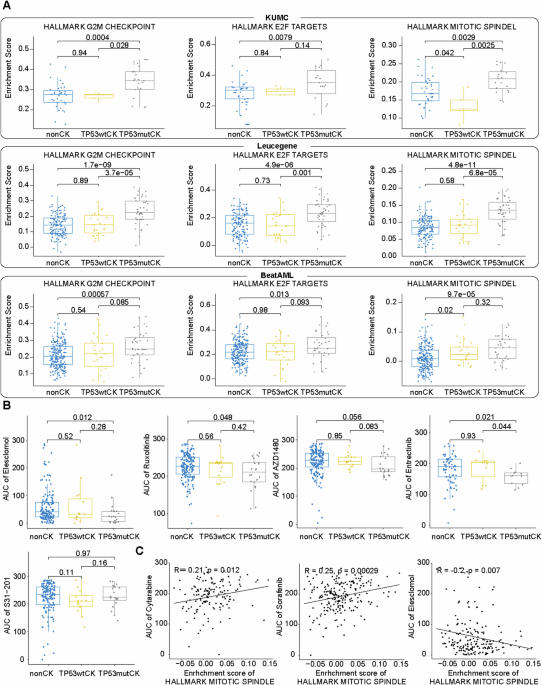
ssGSEA analysis showed that the enrichment scores for CK were significantly higher across all cohorts in pathways involved in the cell cycle (G2/M checkpoint, mitotic spindle, and E2F target), DNA damage (UV response up), heme metabolism, and cholesterol homeostasis (Supplementary Table S3). Compared with non-CK, TP53mut CK showed significantly higher enrichment scores for most of the enriched pathways in CK except for heme metabolism. In contrast, TP53wt CK only showed enrichment in the cholesterol homeostasis pathway (Supplementary Table S4). Among the cell cycle-related pathways, TP53mut CK showed higher enrichment scores compared to TP53wt CK, with statistical significance across all cohorts in the G2/M checkpoint pathway and at least two cohorts in mitotic spindle and E2F target pathways (Fig. 1A). The over-representation analysis of differentially expressed genes also confirmed enrichment of cell cycle-related gene ontology terms in TP53mut CK across all cohorts (Supplementary Fig. S4).
Fig. 1: Comparative analysis of cell cycle-related gene set enrichment scores and the correlation between cell cycle-related pathway and drug response.
A The scaled enrichment scores from single-sample gene set enrichment analysis (ssGSEA) for three cell cycle-related hallmark gene sets (“HALLMARK_G2M_CHECKPOINT,” “HALLMARK_E2F_TARGETS,” and “HALLMARK_MITOTIC_SPINDLE”) were compared across three groups: non-CK, TP53mut CK and TP53wt CK. Each row represents a different cohort, and each column represents a different gene set. B Drug sensitivity of the non-CK, TP53wt CK, and TP53mut CK groups to elesclomol, JAK inhibitors (ruxolitinib, S1312, and AZD1480), and the STAT3 inhibitor (S31-201). Significance was assessed using the Mann–Whitney test. C Scatter plots showing the correlation between the enrichment score of the “HALLMARK_MITOTIC_SPINDLE” gene set and the AUC for three drugs: (left) cytarabine, (middle) sorafenib, and (right) elesclomol.
Ex-vivo drug sensitivity analysis revealed that AML with CK was generally less sensitive than those without CK to most drugs (Supplementary Fig. S5A). Both TP53mut CK and TP53wt CK-AML correlated with resistance to broad range of drugs, however, significantly reduced sensitivities to standard AML treatment agents, such as cytarabine and sorafenib, were noted only in TP53mut CK (Supplementary Fig. S5B, C). In the CK group, TP53 mutations were correlated with sensitivity to elesclomol (P = 0.012), JAK inhibitors (ruxolitinib, P = 0.048; AZD1480, P = 0.056), and entrectinib (P = 0.021) (Fig. 1B). For TP53wt CK-AML, STAT3 inhibitor (S31-201) was the only potent compound that showed a trend towards sensitivity (P = 0.106) (Fig. 1B). We hypothesized that cell cycle-related pathway enrichment may be associated with variations in drug sensitivity and observed a correlation between cell cycle-related pathway enrichment scores and drug sensitivity patterns. The mitotic spindle pathway score was positively correlated with cytarabine and sorafenib AUC (cytarabine: r = 0.21, P = 0.012; sorafenib: r = 0.25, P < 0.001) and negatively correlated with elesclomol AUC (r = −0.20, P = 0.007) (Fig. 1C).
We deconvoluted AML cell populations using bulk RNA-seq data based on the composition of the cellular hierarchy. Compared to non-CK group, the CK samples showed a higher proportion of primitive-dominant hierarchies (44.5% for CK vs. 23.4% for non-CK; P < 0.001) and a lower proportion of GMP-dominant hierarchies (6.5% for CK vs. 19.6% for non-CK; P < 0.001), consistent with previous reports [10, 11], and across all three cohorts (Supplementary Fig. S6). Notably, TP53mut CK had a lower proportion of primitive hierarchies (40.2% vs. 54.2%) and a higher proportion of intermediate and GMP hierarchies than TP53wt CK (9.4% vs. 2.1%), which was consistent across all three cohorts (Fig. 2A and Supplementary Fig. S6).
Fig. 2: Cellular hierarchy composition and its association with drug response, cell cycle related gene set enrichment score, and patient survival.
A Distribution of cellular hierarchy composition (GMP, Intermediate, Mature, and Primitive) across non-CK, TP53wt CK, and TP53mut CK groups. B Pearson correlation between the proportion of each cell type and drug sensitivity (-AUC) in the BeatAML cohort, with colors representing the direction of the correlation. Only significant correlations (P < 0.05) are depicted. C Comparison of AUC values for elesclomol and D enrichment scores of three cell cycle-related gene sets between primitive and other cell types in the TP53mut CK group. Detailed cell types within the ‘Others’ category is shown for the hall mark mitotic spindle gene set. E Overall survival outcomes of AML hierarchy subtypes in the TP53mut CK group. Differences in survival outcomes between the subtypes were assessed using the log-rank test.
In AML without CK, we observed venetoclax sensitivity in primitive-like cell types and resistance in mono-like cell types, which is consistent with previous findings. However, a lack of association between leukemic cell type profiles and venetoclax sensitivity was observed in both TP53mut CK and TP53wt CK-AML (Fig. 2B), suggesting that cell maturation state may not be a reliable predictor of venetoclax sensitivity in CK-AML. In CK-AML, particularly in TP53mut CK, a clear association between cellular hierarchy and drug response was observed in elesclomol. Primitive-like cell types were more sensitive to elesclomol whereas promono-like cell types were more resistant. On analysis of subtypes enriched in cell hierarchy composition and elesclomol AUC, a significantly lower AUC was observed particularly in TP53mut CK with primitive-dominant hierarchies (P = 0.010) (Fig. 2C). Given that the limited efficacy of elesclomol in previous AML trial may have been due to a lack of biomarkers, the findings highlight the potential of cellular composition as a biomarker for predicting elesclomol sensitivity [13, 14].
We further investigated the association between cell cycle-related pathway enrichment scores and dominant cellular hierarchies. The enrichment scores for cell cycle-related pathways were relatively low in the primitive cell type-dominant group (Fig. 2D). The primitive cell type-dominant group showed abundance of quiescent and primed LSPCs but not cycling LSPCs, a population defined by enrichment for CTCF targets and broad enrichment of E2F targets [11]. We then compared the primitive cell type-dominant group of TP53mutCK to those of the non-CK group and found significantly higher mitotic spindle enrichment scores (P = 0.014), confirming the association of TP53 mutation and CK with cell cycle-related pathway enrichment in the primitive cell type-dominant group.
In line with the observed associations with cell cycle-related pathway scores and drug response to cytarabine and sorafenib, we focused our subsequent analysis on the primitive cell type-dominant cellular hierarchy composition group of TP53mut CK. This subgroup of TP53mut CK showed a relatively lower AUC value for cytarabine than the other TP53mut CK subgroups (Supplementary Fig. S7). We analyzed the survival outcomes of this subgroup, and identified a better prognosis for this subgroup than for other cell types among TP53mut CK in the BeatAML cohort (P = 0.011) (Fig. 2E and Supplementary Table S5). Taken together, within the TP53mut CK, the primitive cell type-dominant TP53mut CK subgroup is characterized by lower cell cycle-related pathway enrichment, drug sensitivity patterns such as increased sensitivity to elesclomol and relatively lower resistance to standard treatments, and potentially better clinical outcomes.
In conclusion, this study presents an overview of the genetic profiles of AML/MDS-IB with CK and demonstrates the distinction between TP53mut CK and TP53wt CK in terms of gene mutations, dysregulated pathways, cellular hierarchy composition, and drug sensitivity patterns. Notably, by integrating cell cycle-related pathway scores and cellular hierarchy composition data, we identified a subgroup of TP53mut CK with better prognosis and unique drug sensitivity patterns. The ex vivo drug sensitivity analysis remains preliminary and not independently validated in our cohort. Future research incorporating ex-vivo drug sensitivity analysis along with RNA-seq data, is essential to confirm these associations. Furthermore, integrating TP53 mutation allelic status (single- or multi-hit) would expand the understanding of the genetic profile of TP53mut CK.
留言 (0)