
記住我
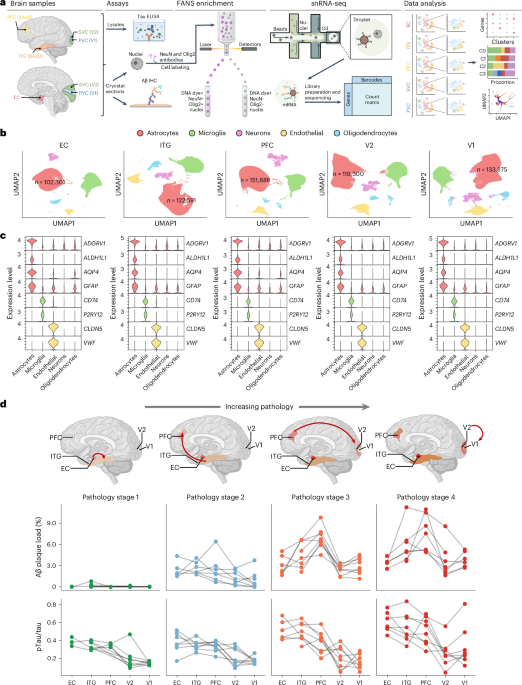

Using an enrichment strategy consisting of depleting NeuN-positive neurons and OLIG2-positive oligodendrocytes via fluorescence-activated nuclei sorting (FANS)18,19 (Fig. 1a and Supplementary Fig. 1a), we obtained a total of 628,943 astrocyte nuclei from five brain regions of 32 individuals with autopsy findings along the normal aging to severe AD neuropathological continuum (Fig. 1a). The five brain regions were selected to represent the hierarchical spreading of pTau NFTs along brain networks as categorized by the Braak NFT staging system20,21 and included entorhinal cortex (EC), inferior temporal gyrus (ITG, Brodmann area (BA) 20), dorsolateral prefrontal cortex (PFC, BA46), secondary (association) visual cortex (V2, BA18/19) and primary visual cortex (V1, BA17). Our strategy to enrich astrocyte nuclei was effective, as indicated by the low numbers of neuronal and oligodendroglial nuclei identified (Fig. 1b). Astrocyte nuclei were identified by the expression levels of the marker genes ADGRV1 (refs. 13,22), ALDH1L1, aquaporin-4 (AQP4) and GFAP (Fig. 1c), resulting in over 100,000 astrocyte nuclei in each brain region with ≈6,000 average total number of reads and ≈2,500 average total number of genes detected, which is orders of magnitude higher than previous studies9,10,11,12,13,14,15 (Supplementary Fig. 1b).
Fig. 1: A molecular survey of astrocytes in five brain regions affected stereotypically in AD.
a, Experimental overview of our snRNA-seq study. The figure was created with BioRender.comb, UMAP visualization showing clustering of NEUN−/OLIG2− nuclei. c, Violin plots illustrate the expression levels of cell type-specific marker genes in NEUN−/OLIG2− nuclei across the five brain regions. d, Results of Aβ plaque load (percentage of immunoreactive area fraction) and pTau/tau ratio (measured by ELISA) in adjacent samples to those used for snRNA-seq across brain regions and pathology stages. SVC, secondary (association) visual cortex; PVC, primary visual cortex; IHC, immunohistochemistry; FANS, fluorescence-activated nuclei sorting.
Because astrocyte morphology changes near Aβ plaques and pTau NFTs23,24, we quantified the local burden of ADNC in adjacent tissue samples by measuring the 3D6-positive Aβ-immunoreactive percentage area fraction via immunohistochemistry and the pTau/total tau ratio via ELISA. To reflect the progression of ADNC, we then grouped the 32 donors into four pathology stages based on their global semiquantitative measures of neuritic plaques (Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuritic plaque (NP) score) and NFTs (Braak NFT stages) complemented with these immunohistochemical and biochemical quantitative measures of local Aβ and pTau burdens. The four pathology stages were as follows: (1) not AD/low ADNC burden (no NPs, Braak NFT stages 0/I/II), (2) intermediate ADNC burden (sparse or moderate NPs and Braak NFT stages II/III), (3) high ADNC burden with moderate or frequent NPs and Braak NFT stage V and (4) high ADNC burden with moderate or frequent NPs and Braak NFT stage VI. We separated the latter two groups because, by definition, the primary visual cortex (V1 region) bears NFTs only in Braak NFT stage VI20,21. Figure 1d illustrates the quantitative measures of ADNC across brain regions of the 32 donors grouped in these four pathological stages. Within each stage, the Aβ plaque load was relatively constant across brain regions except for higher levels in PFC at late stages. By contrast, the pTau/tau ratio was highest in EC and demonstrated the expected pattern EC > ITG > PFC > V2 > V1 across all stages, consistent with the stereotypical hierarchical accumulation of NFTs along neural networks20,21 (Fig. 1d and Supplementary Fig. 1c). The demographic and neuropathological characteristics of the study donors, including quantitative measures of Aβ and pTau, are summarized in Table 1 and detailed in Supplementary Data 1.
Table 1 Donor summary characteristicsRegional heterogeneity of astrocyte transcriptome in the normal aging brainNumerous transcriptomics studies have described the region-specific diversity of astrocytes in the mouse8,25,26,27,28,29,30,31,32, but less is known about the transcriptomic heterogeneity of astrocytes across different cortical areas in the aging human brain. Our eight control donors provided a unique opportunity to examine regional differences in the astrocyte transcriptome in the normal aging brain because these donors had essentially no 3D6-positive Aβ plaques across the five brain regions and very low pTau/tau ratios except for the EC, as expected in Braak NFT stages 0/I/II (Fig. 1d). Thus, we asked whether the astrocyte transcriptome varies across regions in the normal aging brain. To address this question, we integrated a total of 246,464 nuclei from all brain regions of these eight individuals and conducted a differential gene expression analysis of each brain region relative to all other regions (Supplementary Data 2). Remarkably, the EC and V1 regions had the highest number of differentially expressed genes (DEGs; EC: 137 up and 152 down; V1: 93 up and 122 down; see Venn diagrams in Fig. 2a). A heatmap with relevant DEGs per region is shown in Fig. 2b. Among other genes of interest, APOE, APP, AQP4 and GJA1 were upregulated, whereas ALDH1L1, LRP1B, microtubule-associated protein tau (MAPT) and SLC1A2 were downregulated in EC relative to the other brain regions. Conversely, CLU, GFAP and MAOB were upregulated, whereas AQP4 and GJA1 were downregulated in V1 versus all other brain regions. Validation with immunohistochemistry confirmed the regional differences between EC and V1 for AQP4 and GJA1 at the protein level (Fig. 2c and Supplementary Figs. 2 and 3).
Fig. 2: Regional heterogeneity of astrocyte transcriptome in the normal aging brain.
a, Venn diagrams show the DEGs—upregulated in red and downregulated in blue—in each brain region relative to all other brain regions in n = 8 donors at pathology stage 1 (no neuritic plaques and Braak NFT stages 0/I/II). b, Heatmap depicts the DEGs in each brain region (upregulated in red and downregulated in blue) ranked by average log FC. c, Fluorescence immunohistochemistry of AQP4 and connexin-43 (GJA1) in EC and V1. Automated immunohistochemistry with the peroxidase-DAB method followed by quantification of AQP4- and GJA1-immunoreactive percentage area fraction in EC and V1 formalin-fixed paraffin-embedded (FFPE) sections from n = 11 pathology stage 1 donors (including two donors from this snRNA-seq study) demonstrated a higher expression of these two proteins in EC versus V1, in agreement with the snRNA-seq results. *P = 0.05, **P = 0.0036, two-sided paired t test. Scale bars—EC, 1 mm; V1, 5 mm; insets, 500 μm. d, Volcano plot illustrates the correlation analysis between pTau/tau ratio and astrocyte gene expression levels in pathology stage 1 donors. The x axis represents the correlation coefficient β, while the y axis indicates the FDR (expressed as −log10(FDR)). Genes in red are positively correlated, and those in blue are negatively correlated at an FDR < 0.05 (−log10(FDR) > 1.3), whereas genes in gray were statistically NS. e, Venn diagrams show the number of genes upregulated in EC (EC high) and/or positively correlated with the pTau/tau ratio (top) and the number of genes downregulated in EC (EC low) and/or genes negatively correlated with pTau/tau ratio (bottom). Genes in each intersection are listed in the boxes. NS, not significant.
These results suggest some degree of region-specificity of astrocyte transcriptome in the allocortex (EC) versus primary neocortex (V1) versus association neocortex (ITG, PFC and V2). To further examine this possibility, we asked whether EC DEGs are driven by the EC microenvironment (that is, allocortex versus neocortex) or by the amount of local EC pTau pathology already present in Braak stages I and II donors (Fig. 1d and Supplementary Data 2). First, we identified a gene set associated with the local pTau burden by regressing gene expression levels against the pTau/tau ratio across all five brain regions in these eight donors (148 positively and 433 negatively correlated genes, false discovery rate (FDR) < 0.05; Supplementary Data 3 and volcano plot in Fig. 2d). Then, to disambiguate EC-specific versus pTau-driven genes, we overlapped these pTau-correlated genes with those enriched in the EC (see Venn diagrams in Fig. 2e). Only 18 of the 137 EC-upregulated genes were positively correlated with the pTau/tau ratio (notably including cytoskeletal genes such as MAP2 and the master transcription factor regulating cholesterol metabolism, SREBF1), whereas only 37 of 152 EC-downregulated genes were negatively correlated with the pTau/tau ratio (including extracellular matrix/glycosaminoglycan metabolism genes, such as LARGE1 and PTN). Taken together, these results support the existence of an allocortex-specific gene expression profile in EC astrocytes, which is distinct from a pTau signature.
Astrocyte transcriptomic changes along the stereotypical spatial progression of ADNext, we examined whether astrocyte gene expression changes track with the stereotypical spatiotemporal progression of AD neuropathology. The present study design with 32 donors along the normal aging to severe AD continuum and five sites that are hierarchically involved in stereotypical AD enabled us to address the hypothesis that the magnitude of astrocyte transcriptomic changes would parallel both the regional vulnerability of neural networks to ADNC (spatial progression) and the temporal accrual of ADNC in a given brain region (temporal progression).
First, to determine whether astrocyte transcriptomic changes follow the neural network predilection of AD progression, we rank ordered the five brain regions based on their known vulnerability to neurofibrillary degeneration20,21 (that is, EC > ITG > PFC > V2 > V1) and performed a differential expression analysis comparing each node of the network with the next (that is, EC versus ITG, ITG versus PFC, PFC versus V2 and V2 versus V1), including all 32 donors and controlling for within-donor correlations. The resulting 504 DEGs between any two adjacent network nodes were then grouped into six spatial gene sets following distinct trajectories along the neural network (Fig. 3a and Supplementary Data 4). Two spatial gene sets changed their expression level monotonically along the network, either decreasing from EC to V1 (gene set 1) or increasing from EC to V1 (gene set 2). Two other spatial gene sets had relatively stable expression levels across all brain regions, except for a peak of upregulation (gene set 3) or downregulation (gene set 4) at the PFC. The last two spatial gene sets exhibited relatively constant expression levels in EC, ITG and PFC, but either decreased (gene set 5) or increased (gene set 6) from the PFC to V2 and V1. Thus, this analysis suggested a significant association between astrocyte gene expression and relevant regions that are hierarchically affected by the AD pathophysiological process. A heatmap depicting the differential expression of the individual genes comprising these spatial gene sets across brain regions at the individual donor level is shown in Supplementary Fig. 4a, demonstrating remarkable consistency with the average profile of these gene sets shown in Fig. 3a. Donor-level expression of representative genes for each spatial trajectory is shown in Supplementary Fig. 4b.
Fig. 3: Astrocyte transcriptomic changes along the stereotypical spatial progression of AD.
a, Spatial trajectory gene sets resulting from clustering the n = 504 DEGs between any two adjacent nodes of the AD network from EC to V1. The y axis is the standardized gene expression, with gray lines representing individual genes, whereas the colored lines represent the mean trend. b, The first two vertical bars represent the association between the average expression of each spatial trajectory gene set in each donor and their pTau/tau ratio and Aβ plaque burden (red indicates statistically significant positive correlation; blue indicates statistically significant negative correlation; gray indicates NS). The last two vertical bars illustrate the results of an overlap test between each spatial trajectory gene set and the region-specific EC-high/EC-low and V1-high/V1-low gene sets derived from normal controls in Fig. 2 (red indicates statistically significant overlap with region-specific upregulated gene set; blue indicates statistically significant overlap with region-specific downregulated gene set; gray indicates NS). c, Functional characterization of each spatial trajectory gene set via pathway analysis; some relevant genes of each pathway are displayed on the right. ROS, reactive oxygen species.
Previous transcriptomic studies comparing AD mouse models have reported similarities and differences in astrocyte responses to Aβ plaques versus pTau NFTs5,7,33. To test whether the spatial trajectories of astrocyte gene expression are also related to local Aβ and pTau levels, we correlated the average standardized expression of each of these spatial gene sets for each brain region and donor with the Aβ plaque burden and the pTau/tau ratio measured in the same brain regions and donors, while controlling for within-donor correlation (Fig. 3b and Supplementary Fig. 4c). Interestingly, the gene set decreasing linearly from EC to V1 (gene set 1) was positively correlated with the pTau/tau ratio, whereas the one increasing from EC to V1 (gene set 2) was negatively correlated with the pTau/tau ratio, suggesting that these two gene sets contain a pTau-associated signature. Gene set 1 contained synapse-associated (GRIA1, GRIP1, KCND2, KCNE4, KCNN3, SYNPO2 and SYTL4), cell–cell communication (APP, AQP4, CNTN1, IL33, ITGB4 and TJP2), cytoskeleton (GSN, MAP2, MAP7, MYBPC1, TLN2 and TTN), extracellular matrix (ADAMTSL3, COL21A1, LAMA4, SERPINI2 and VCAN) and some trophic factors (ANGPT1, EGF, NRP2 and NTRK2), metal binding (CP and FTH1) and lipid metabolism (ABCA1 and LRAT) genes, whereas gene set 2 included genes involved in glutamate neurotransmission (GLUL, GRIA2, NRXN1 and SLC1A2) and extracellular matrix (ADAM12, FREM2, MGAT5 and PTN; Fig. 3c).
By contrast, the gene set specifically upregulated at the PFC (gene set 3) was only positively correlated with the Aβ plaque burden, which was highest in this brain region (Fig. 1d), suggesting that this is an Aβ-associated signature. This gene set was enriched in synapse-associated (DISC1, GRIA4, GRID2 and SYNDIG1), cell–cell communication (APBB1IP, EFNA5, EPHA6 and SPP1) and cytoskeletal (SYNM and FMN1) genes that were distinct from those associated with the pTau/tau ratio (Fig. 3c). On the other hand, the gene set specifically downregulated in the PFC (gene set 4) contained important genes involved in cytoskeleton (ACTB, CAP2, GFAP, MAP1B and VIM), lipid metabolism (APOE, CLU and DGKB) and calcium homeostasis (CALM2, CNN3, RTN1, S100A1 and S100B), as well as many extracellular matrix (CST3, KAZN, HS6ST3, LAMA2, MATN2, SDC4, SERPINA3, SERPINH1, SPARC and TNC), proteostasis (BAG3, CRYAB, DNAJA1, DNAJB1, HSP90AA1, HSPA1A, HSPA1B, HSPB1, HSPB8, HSPD1, HSPE1, HSPH1, UBB and UBC) and antioxidant defense genes (PRDX1, SOD2 and the metallothioneins MT1E, MT1F, MT1G, MT1M, MT1X, MT2A, MT1M, MT2A and MT3; Fig. 3c). While this gene set was negatively correlated with both Aβ plaque burden and pTau/tau ratio, the statistical significance was lost after controlling for brain region.
Finally, spatial gene set 5 was positively correlated with both pTau/tau ratio and Aβ plaque burden, whereas gene set 6 was negatively correlated with both measures, likely representing ADNC-associated pan-reactive upregulated and downregulated signatures, respectively. The pan-reactive genes positively correlated with pTau and Aβ levels included many genes involved in G-protein-coupled receptor (GPCRs) signaling (ARHGEF12, GNA14, GNAQ, PDE3B, PDE4D, PDE4DIP, PDE5A and PRKG1) and intracellular transport (CPQ, DNAH7, DNM3, RANBP3L, SLC1A3, SLC15A2, SLC44A3 and SLCO1C1), suggesting activation of second messenger-mediated signaling cascades and intracellular trafficking of solutes and vesicles (Fig. 3c). Remarkably, the pan-reactive genes negatively correlated with pTau and Aβ levels were predominantly related to cell motility (ACTN, ASAP3, CXCL14, DAAM2, MYH9 and SERPINE2) and metabolism, including glucose (ALDH1A1, DPP6, GAPDH, GYS1, LDHB and MIDN), lipid (ABHD3, OSBPL3 and SLC27A1) and nucleotide (DHFR) pathways, suggesting a failure of energy metabolism in reactive astrocytes associated with chronic exposure to both pTau and Aβ (Fig. 3c).
We then asked whether this association could be driven by the regional differences observed in normal control donors or whether it was mainly explained by the accumulation of Aβ and/or pTau in those brain regions. To test the first possibility, we compared the EC-specific and V1-specific signatures obtained from normal controls (Fig. 2) and these spatial trajectory gene sets derived from the entire sample and, indeed, observed a significant overlap (Supplementary Fig. 4d). Specifically, the gene set with monotonic decrease from EC to V1 (gene set 1) was significantly enriched in EC-high and V1-low genes, whereas the gene set with monotonic increase from EC to V1 (gene set 2) was significantly enriched in EC-low and V1-high genes. The gene set specifically upregulated in PFC (gene set 3) was not enriched in either EC or V1 signatures, whereas the gene set specifically downregulated in PFC was enriched in EC-high and V1-high genes. The gene set with relatively stable expression in EC through PFC and lower expression in V2 and V1 (gene set 5) was enriched in EC-low and V1-low genes, and the gene set with stable levels in EC through PFC but increase in V2 and V1 (gene set 6) was expectedly enriched in V1-high genes (Fig. 3b). These data suggest that some of the regional variations in astrocyte transcriptome along the AD neural network are already present in the normal aging brain, possibly driven by exposure of astrocytes to microenvironmental factors particular to each brain region.
Taken together, these results indicate that the astrocyte gene expression profile parallels the typical spatial progression of AD pathology along neural networks and suggest that these astrocyte transcriptomic changes are partly associated with the local levels of Aβ plaques and/or pTau and partly explained by region-specific microenvironmental factors.
Astrocyte transcriptomic changes along the temporal progression of ADOnce we established the spatial progression of astrocyte transcriptomic changes in the normal aging to severe AD continuum, we sought to determine whether astrocyte transcriptomic changes also parallel the temporal accrual of ADNC within a given brain region. To this end, we conducted a differential expression analysis comparing each of the aforementioned four pathology stages with the next while controlling for brain region and within-donor correlation. We decided to separate Braak stages V and VI donors to investigate possible end-stage changes in astrocyte transcriptome and because, by definition, the primary visual cortex (V1) only contains NFTs in Braak stage VI donors but is spared from NFTs in Braak stage V donors20,21. The resulting 798 DEGs between any two adjacent stages were, thus, considered temporally associated with AD pathophysiology. These 798 DEGs were grouped into six different temporal gene sets with distinct temporal trajectories and variable strength of association with the local Aβ and pTau levels (Fig. 4 and Supplementary Data 5).
Fig. 4: Astrocyte transcriptomic changes along the temporal progression of AD.
a, Temporal trajectory gene sets resulting from clustering the n = 798 DEGs between any two adjacent pathology stages from early to end stage. The y axis is the standardized gene expression, with gray lines representing individual genes, whereas the colored lines represent the mean trend. b, Functional characterization of each temporal trajectory gene set via pathways analysis; some relevant genes of each pathway are displayed on the right. MAPK, mitogen-activated protein kinase; PPARα, peroxisome proliferator-activated receptor α.
One set (temporal gene set 1) had the highest expression at an early stage (no NPs, Braak NFT stages 0/I/II) and then decreased and remained relatively low along all the other stages, suggesting that it corresponds to a homeostatic signature of astrocytes. This gene set included many trophic factors and their receptors (ANGPTL4, APC, EGLN3, FGF2, GFRA1, HGF, IGF1R, NRP1, NTRK3, TGFB2, TGFBR3 and VEGFA), extracellular matrix (GPC4, MATN2, SERPINA3, SERPINE2 and SPOCK1), cytoskeleton (ACTN1, FIGN, MACF1, MAPRE2, MAST4 and MYH9) and some metal binding (CP, FTL and MT2A), antioxidant (MGST1 and MSRA) but also pro-oxidant enzymes (MAOB), neuroinflammation (MAP3K14, MAP4K4, OSMR, SOCS3 and TAB2), calcium homeostasis (CALN1, CNN3 and S100A1) and phagocytosis (LGALS3 and SCARA3) genes.
Another gene set (gene set 2) peaked at the intermediate ADNC stage (sparse/moderate NPs and Braak NFT stage II/III) and then returned to baseline at late and end stages, suggesting a transient activation of this gene set in response to the initial accumulation of ADNC. This early gene program included trophic and survival factors (FGF1, FGF14, HIF1A and NRP2), extracellular matrix (GPC6, KAZN, LAMA4 and TNC), cytoskeleton (ABI1, CLIP1, MYO1E and VCL) and neuroinflammation (CHI3L1, IFI16, IL1R1, IL6R and RCAN1) genes, which differ from those predominant in the early ADNC stage. It also encompassed genes involved in lipid metabolism (ACSL3, ELOVL5, ELOVL6, LDLR, LEPR, LPIN1, NCEH1, OSBPL6, PLA2G4C, PLCL1, PLPP3 and SGMS2), glycosaminoglycan metabolism (EXT1, GALNT2, MAN2A1, ST6GALNAC3, SULF1 and UGP2), synapse-associated (GRIK2, PCLO, STXBP6 and SYTL2) and GPCR-mediated signaling (GPR158, KALRN, PDE4B, PDE4D, RASGEF1B, RGS6 and TRIO).
Two other temporal gene sets either peaked (gene set 3) or dropped (gene set 4) at the late stage (moderate/frequent NPs, Braak NFT stage V) but surprisingly returned to near baseline at end stage (moderate/frequent NPs, Braak NFT stage VI), suggesting a vigorous response of astrocytes to nearby Aβ plaques and NFTs that they ultimately cannot sustain and turn off as this exposure becomes chronic (that is, response exhaustion). The upregulated late gene program comprised genes involved in proteostasis/response to heat stress (AHSA1, BAG3, CCT4, CRYAB, DNAJA1, DNAJB1, DNAJB6, HSP90AA1, HSP90AB1, HSPA1A, HSPA1B, HSPA4, HSPA4L, HSPA8, HSPA9, HSPB1, HSPD1, HSPE1, HSPH1, MIB1, OTUD7B, SQSTM1, ST13, UBB, UBC, UBE2B and UBE2E2), protein translation (EEF1A1, EIF1, EIF2S2, RPL37A, RPL38, RPLP2, RPS21, RPS24 and RPS28), energy metabolism (ENO1, GAPDH, LDHB and PGK1), mitochondrial electron transport chain (ATP5F1E, ATP5MD, ATP5ME, ATP5PF, COX6A1, COX6C, COX7C, NDUFA4, NDUFB2, NDUFB4, NDUFC1 and UQCRB), antioxidant defense (NFE2L2, NXN, PRDX1, SOD1, SOD2 and the metallothionein genes (MT1E, MT1F, MT1G, MT1M, MT1X and MT3)), neuroinflammation including interferon response (IFITM2, IFITM3, IL17RB, IRS2, NFAT5, PTGES3 and STAT3), cell–cell communication (CD59, CDH23, CHL1, CNTNAP2, CNTNAP3, CNTNAP3B, FGFR1 and RGMA) and some genes involved in lipid metabolism (ABHD3, CLU, OAZ1 and OSBPL1A), cytoskeleton (CLIP2, MAP2, SYNM and VIM), extracellular matrix (MMP16, PLOD2, PLOD3, SERPINH1, SPP1 and ST6GALNAC6), synaptic function (CAMK2D, CAMK2N1, FOS, GAB1, NRXN3 and RIMS1) and intracellular transport/trafficking (ATP2C1, CPE, DYNLRB1, SLC7A5, SLC7A11, SLC9B2, SLC20A2 and SNX3). The downregulated late gene program included genes involved in glutamate neurotransmission (GLUD1, GRIA2 and SLC1A3), extracellular matrix (COL28A1, CSGALNACT1, EPM2A and GPC5) and intracellular transport (GOLGA8B, SLC4A4, SLC13A3 and TVP23C). Interestingly, many of these late-stage genes overlap with the Aβ/pTau-unrelated genes (gene set 4) of the spatial progression analysis. This lack of correlation with Aβ and pTau is likely explained by the apparent normalization of their expression levels in end-stage disease (Braak stage VI) despite the further accumulation of Aβ and especially pTau in all brain regions of Braak stage VI donors (Fig. 1d).
Moreover, we identified a smaller set (gene set 5) whose expression levels remained relatively stable throughout early, intermediate and late stages and only increased at the end stage (moderate/frequent NPs, Braak NFT stage VI). This gene set contained glutamate metabolism (GLUL and SLC1A2), extracellular matrix (ADAMTSL3, COL21A1, HPSE2, SDC4 and VCAN), lipid metabolism (ACACB, ACSS1, DGKG, OSBPL11, PHYHD1, PHYHIPL, PLCE1, PPARGC1A and SREBF1), trophic factors (FGFR3 and NTRK2), cell–cell communication (CDH20, CLEC16A, CNTN1, ITGB4 and NLGN4Y) and intracellular transport (DYNC2H1, SCLT1, SLC14A1, SLC18B1, SLC24A4 and SLC44A1) genes. Finally, gene set 6 had low expression at early and intermediate stages, peaked at the late stage and decreased at the end stage without returning to baseline; relevant genes pertained to proteostasis (CTSD, DNAJB2, HSPB8 and NEDD4L), energy metabolism (ALDH2, CKB and PFKP), extracellular matrix (B4GALNT4, COL5A3, COL27A1, CST3, FLRT2, PLEC and PLXNB1), intracellular transport and trafficking (ATP2B4, SLC27A1, SLC38A2, SLC39A11, SLC39A12 and TRAK1), cell–cell communication (GJA1), lipid metabolism (APOE and LRP4) and nuclear receptors (RORA and RXRA).
Compared to the spatial gene sets, the heatmap showing the differential expression of the individual genes comprising these temporal gene sets across pathology stages at the donor level showed slightly higher interdonor variability (Supplementary Fig. 4a), possibly reflecting differences in the duration of each stage across donors. Donor-level expression of representative genes for each temporal trajectory is shown in Supplementary Fig. 4b. Interestingly, there was minimal overlap between the spatial and temporal gene sets (Supplementary Fig. 4e), suggesting that a complex interplay of intrinsic properties of the regional microenvironment, local amounts of Aβ and pTau and duration of the exposure to Aβ and pTau insults governs astrocyte gene expression. Indeed, these results indicate that both brain region and Braak NFT stage should be carefully considered when comparing the astrocyte transcriptome of AD versus normal control brains because astrocyte transcriptomic responses to ADNC might be attenuated or exhausted in end-stage disease (Braak NFT stage VI) in severely and chronically affected brain areas.
Astrocyte clustering analysis reveals diverse transcriptomic programs across brain regions and pathology stagesWhile two broad categories of astrocytes, homeostatic and reactive, have been traditionally postulated, the above-mentioned results suggested a more complex picture of astrocyte reactivity, as put forward in a recent consensus statement on reactive astrocytes1. We asked whether distinct subpopulations or states can be distinguished based on their gene expression programs. To address this question, we randomly sampled 500 nuclei from each brain region of each donor and integrated and clustered these nuclei into nine distinct astrocyte subclusters (Uniform Manifold Approximation and Projection (UMAP) in Fig. 5a). Two small clusters had genes typically found in other cell types, specifically neurons (astNeu) and microglia (astMic), and were considered hybrids or doublets (although their estimated doublet score was very low). Inspection of both the top DEGs defining the subclusters (that is, subcluster marker genes available in Supplementary Data 6) and the enriched pathways revealed an interesting functional specialization of the remaining seven subclusters. The subcluster astH0 was enriched in glutamatergic (GLUL, GRIA2, GRM3 and SLC1A2) and gabaergic (SLC6A1 and SLC6A11) neurotransmission genes, along with potassium-buffering channels (KCNJ10 and KCNJ16) and cell adhesion molecules involved in astrocyte–astrocyte (GJA1) and astrocyte–neuron interactions (ERBB4 and NRXN1), consistent with a canonical protoplasmic homeostatic phenotype (Fig. 5b,c). By contrast, astR1 and astR2 were enriched in GFAP, S100B, HSF1-mediated stress response (CRYAB, HSPA1B, HSPB8 and UBC), extracellular matrix (CD44, COL21A1, GPC6, LAMA4, SERPINA3, TNC and VCAN) and oxidative stress (MAOB), but differed in some other marker genes (for example, ADAMTSL3, AQP1, C3, MAP1B, MAPT and SOD2 in astR1 versus GRIA1 and SPARC in astR2), suggesting that they are different states of astrocyte reactivity (Fig. 5b,c). Subcluster astR0 also exhibited a reactive phenotype and was further enriched in extracellular matrix genes (COL21A1, COL24A1, COLGAT and LAMA2). Fluorescence immunohistochemistry validated some of these homeostatic and reactive markers in postmortem brain sections from control and AD donors, and a spatial quantitative analysis centered in thioflavin-S-positive dense-core Aβ plaques demonstrated a gradient of expression of the reactive markers from the plaque to the peri-plaque halo (≤50 μm) to areas distant (>50 μm) from plaques in the AD cortex, whereas no effect of sham plaques was observed in control cortex (Fig. 5d,e and Supplementary Figs. 5–7). Fluorescence in situ hybridization via RNAScope confirmed the expression of MAPT by astrocytes (Supplementary Fig. 8).
Fig. 5: Clustering reveals homeostatic, reactive and intermediate astrocytes.
a, UMAP plot of a subsample of 500 astrocyte nuclei from each donor and region, with the colors representing each of the ten astrocyte subclusters resulting from the clustering. b, Bubble plots illustrate the expression z scores of selected marker genes defining the nine astrocyte subclusters. c, Characterization of astrocyte subclusters with pathway enrichment analysis. Bar plots represent the statistical significance (−log10(FDR)) of the functional pathways defining the main astrocyte subclusters. d, Double fluorescence immunohistochemistry for selected markers with thioflavin-S (ThioS) counterstaining in FFPE sections from the temporal association cortex of control (CTRL) and AD donors show reduced expression of some homeostatic markers and increased expression of some reactive markers in GFAP+ astrocytes surrounding some ThioS+ Aβ plaques. Note that representative photomicrographs from at least three CTRL and three AD donors were taken with similar exposure time and display settings for appropriate comparison. Scale bar = 10 μm. e, Violin plots show a statistically significant or marginally significant higher expression of reactive astrocyte markers in AD (pathology groups 3 and 4, n = 7) versus CTRL (pathology group 1, n = 6) centered within and around ThioS+ Aβ plaques. Individual data points are shown as dots, and horizontal lines represent the median value. Note that the gradient of expression of these reactive astrocyte markers from ThioS+ Aβ plaques to the plaque vicinity (≤50 μm) and to plaque-free distant areas (>50 μm from nearest plaque edge) in the AD temporal neocortex, while no such gradient is observed relative to sham plaques from CTRL cortex. Data from AD donors correspond to n = 50 randomly selected ThioS+ Aβ plaques per donor distributed throughout all layers of a temporal neocortex tissue section, their 50 μm halo and n = 50 distant (>50 μm) ROIs of similar size per donor. For CTRL donors, n = 50 sham plaques of similar size per donor, their 50 μm halo and n = 50 ROIs of similar size located far from them (>50 μm) per donor were analyzed. Data were analyzed running mixed-effects models with diagnosis and location as fixed effects and donor ID as random effect to account for within-donor correlation. FGFR, fibroblast growth factor receptor.
留言 (0)