
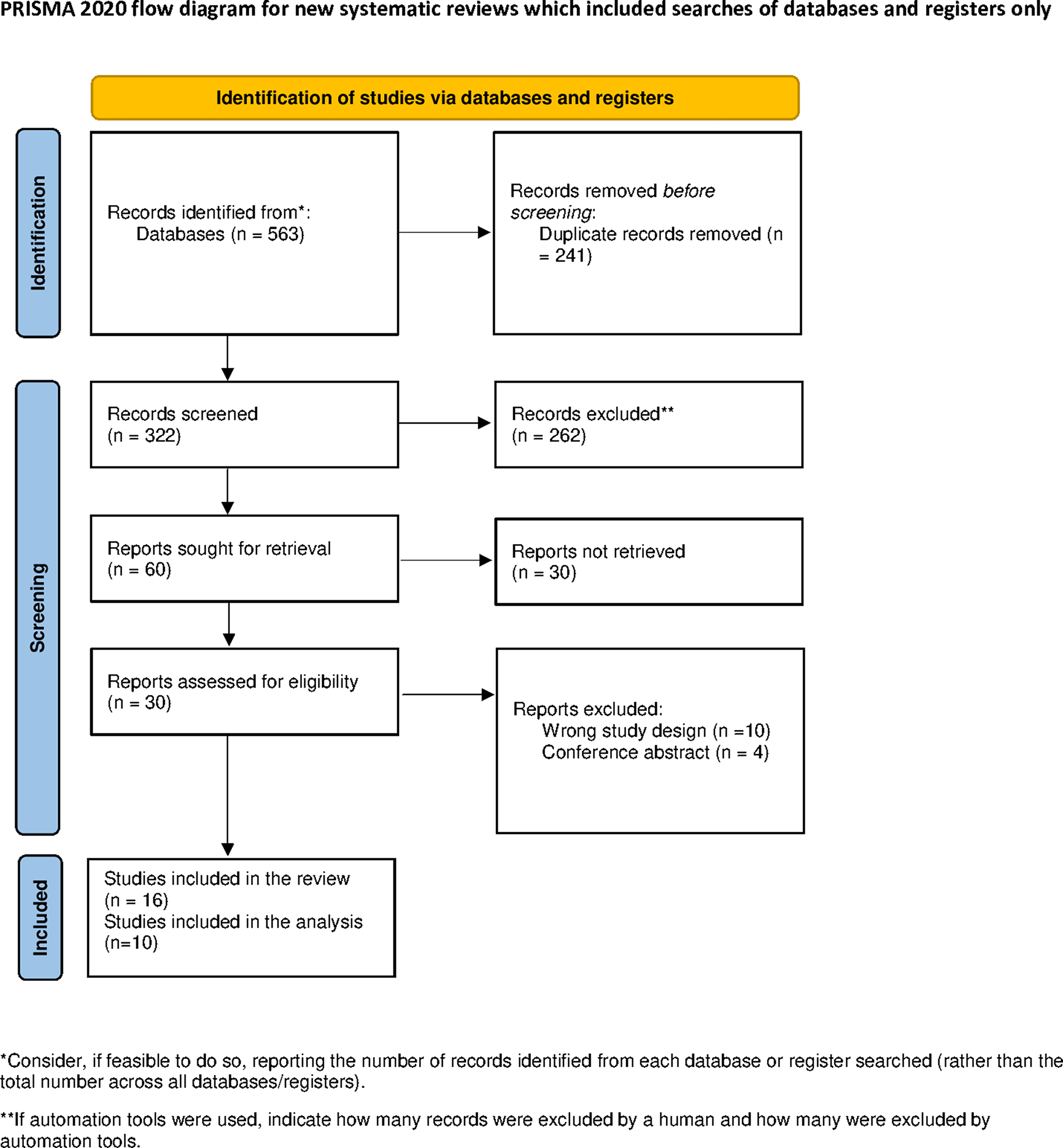
記住我
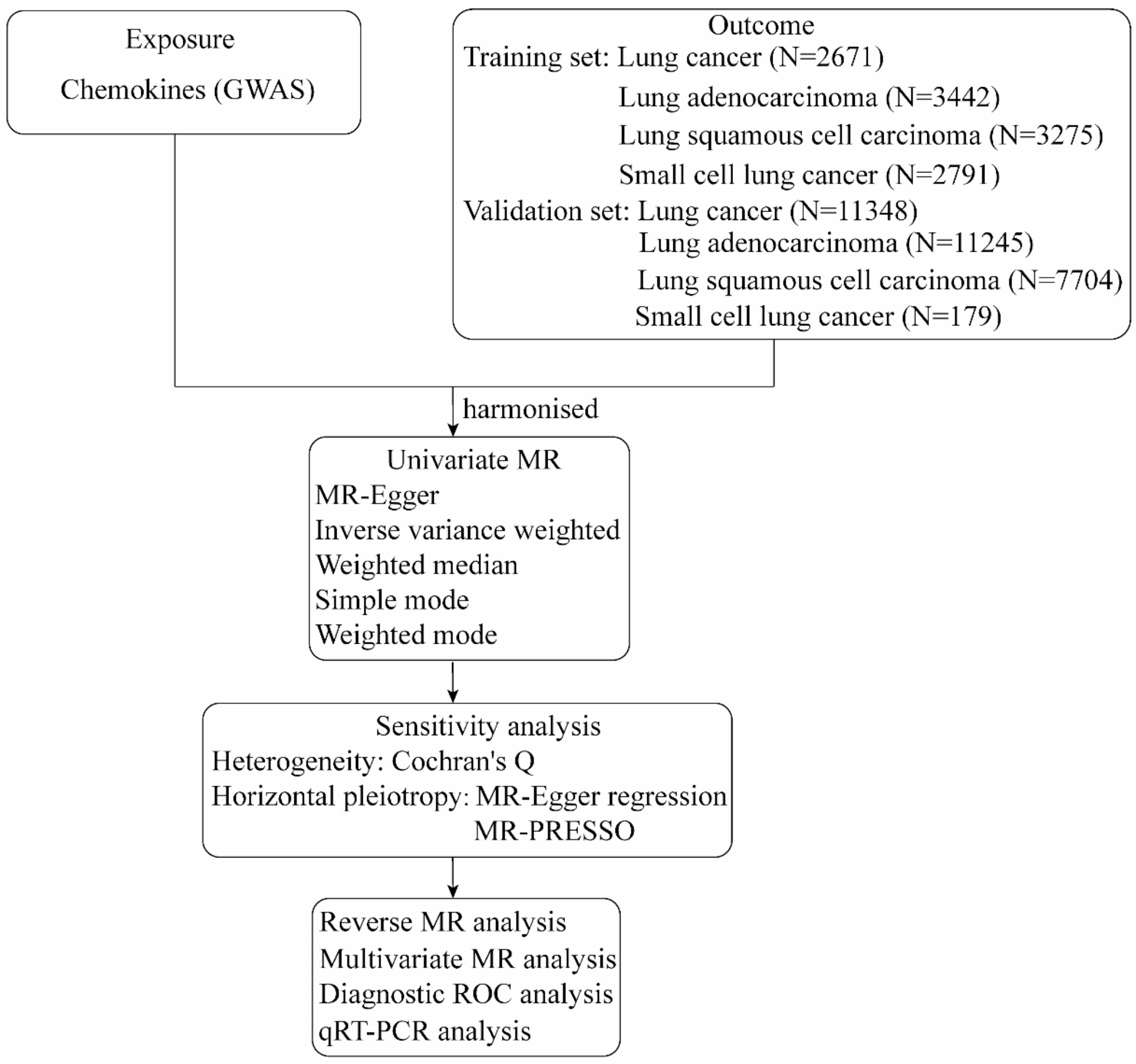




In this study, we used GWAS-derived chemokines as instrumental variables (IVs) to investigate the causal effects of chemokines on LC using two-sample MR [14]. The positive results were validated with additional LC samples. Subsequently, the effect of chemokines mediated by smoking behavior (including ever smoker, current cigarette smokers, light smokers, cigarettes smoked per day) on LC was assessed. Finally, it predicted the accuracy of the discovered chemokines to diagnose LC. A graphical overview of the study design is shown in Fig. 1.
Fig. 1
A graphical overview of the study design
Data sources and instrumental variablesIVs for chemokines were determined from public GWAS summary statistics and did not require individual-level data [15]. After literature search and GWAS database screening, we identified 38 chemokines. GWAS summary data for LC were obtained from UK Biobank, Transdisciplinary Research in Cancer of the Lung (TRICL) and International Lung Cancer Consortium (ILCCO) [16]. All participants were of European descent. Association analyses were performed for lung cancer (2671 cases, 372016 controls), lung adenocarcinoma (LUAD) (3442 cases, 14894 controls), lung squamous cell carcinoma (LUSC) (3275 cases, 15038 controls), and small cell lung cancer (SCLC) (2791 cases, 20580 controls). Validation analyses were performed in another dataset, including lung cancer (11348 cases, 15861 controls), lung adenocarcinoma (11245 cases, 54619 controls), lung squamous cell carcinoma (7704 cases, 54763 controls), and small cell lung cancer (179 cases, 174006 controls). The TCGA-LUAD and TCGA-LUSC datasets were downloaded from the UCSC Xena database (https://xena.ucsc.edu), and the expression matrix of the GSE149507 small cell lung cancer dataset was downloaded from the GEO website (https://www.ncbi.nlm.nih.gov/geo/).
Genome-wide single-nucleotide polymorphisms (SNPs) associated with chemokines were generated from the full-site significance threshold P < 1.0 × 10− 8 [14, 17]. However, CCL1, CCL13, CCL20, CXCL9, CXCL10, CCL13, and CXCL14 had no associated SNPs, so the threshold P < 5.0 × 10− 6 was chosen. The following quality control steps were used to select SNPs to ensure the stability and accuracy of the causal association results between chemokines and LC risk. (1) Excluding SNPs with allelic inconsistency between the exposure and outcome samples (such as A / C); (2) Removing palindromic SNPs; (3) Eliminating SNPs causing linkage disequilibrium by employing the PLINK clumping method (r2 < 0.001 and clump window = 10,000 kb); (4) Removing SNPs with minor allele frequency (MAF) < 0.01; (5) Using the PhenoScanners V2 database (phenoscanner.medschl.cam.ac.uk) [18], removal of mixed exposure and outcome related SNPs, such as two SNPs-rs508977 and rs1973612 of growth-regulated protein alpha levels were associated with exposure, thus excluding growth-regulated protein alpha levels. Abnormal SNPs were detected using MR-pleiotropy residual sum and outlier (MR-PRESSO) and pleiotropy was eliminated by removing outliers, followed by MR analysis after removing SNPs with pleiotropy. The R2 and F statistics were used to assess weak instrumental deviation [19], and the F statistic was calculated using this formula: R2 (n-k-1) / k (1-R2). Where n represents the number of samples, k represents the number of IVs, and R2 represents the variance explained by the IVs.
MR analysisIn this study, five methods, random or fixed effects inverse variance weighted (IVW) [20], weighted median estimation (WME) [20], MR-Egger [12], Simple mode (SM) [21], and Weighted mode (WM) [21], were used to verify whether there is a causal association between chemokines and lung cancer. IVW method, which uses the Wald ratio method for the association of individual SNP followed by meta-aggregation of multiple locus effects using a choice of fixed or random effects models, is able to provide the most accurate estimates of effects [20]. The premise of the WME method is to give an accurate assessment based on the assumption that at least 50% of IVs are valid [20]. MR-Egger method takes into account the presence of an intercept term that detects and adjusts for horizontal pleiotropy, and if horizontal pleiotropy is not present, then MR-Egger regression and IVW results are essentially identical [22]. SM and WM methods are also two important statistics in MR analysis. In this study, the IVW method was used as the main method, and other methods were used as supplements.
Sensitivity analysisTo further test the accuracy and stability of the findings, we performed sensitivity analysis using heterogeneity test, horizontal multiple validity test, and leave-one-out method. Cochrane’s Q test was used to assess the heterogeneity of each chemokine-associated SNPs; if heterogeneity existed (P < 0.05), a randomized IVW method was used; if heterogeneity did not exist (P > 0.05), a fixed IVW method was used. The MR-Egger method means that when the intercept term is very different from 0, it indicates the presence of horizontal pleiotropy, in which case it is necessary to eliminate the horizontal pleiotropy by removing the abnormal SNPs with MR-PRESSO and re-performing the MR analysis [23, 24]. The leave-one-out method was used to assess whether MR causality was driven by a single SNP. To avoid false positive results, the false discovery rate (FDR) adjusted p-value in the main analysis was calculated using the fdrtool package [25]. Reverse MR analysis of chemokines causally related to LC in the forward MR analysis. Considering that smoking is a risk factor for LC, we performed a multivariate Mendelian randomization (MVMR) analysis to estimate the effect of each chemokine on LC after adjusting for smoking status. Further predicting the accuracy of the discovered chemokines to diagnose LC, we performed sensitivity and specificity analysis using expression matrices from the TCGA and GEO data.
Lung adenocarcinoma cell linesHBE, A549, H1299, H1975, and PC9 cell lines were acquired from the China Cell Resource Center (Shanghai, China). The cells were cultured in RPMI 1640 (Solarbio) supplemented with 10% fetal bovine serum (Procell). The cells were then incubated in a humidified incubator (Thermo Scientific, China) with 5% CO2 at 37 °C.
RNA extraction and quantitative real-time PCR (qRT-PCR) assayRNA extraction from cells was performed using the TRIzol reagent (Vazyme). The cDNA was synthesized using the Hifair® III 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus) (Yeasen Biotechnology, Shanghai, China). The quantitative real time PCR was carried out with Hieff®qPCR SYBR Green Master Mix (High Rox Plus) (Yeasen Biotechnology, Shanghai, China) in the StepOne Plus Real-Time PCR System (Applied Biosystems). The relative expression levels of CCL21 mRNA were normalized to GAPDH as endogenous control respectively by using the 2−△△ct method. The primer sequences were as follows: CCL21: F: 5ʹ- GTTGCCTCAAGTACAGCCAAA-3ʹ, R: 5ʹ- AGAACAGGATAGCTGGGATGG-3ʹ; GAPDH: F: 5ʹ-TCATTTCCTGGTATGACAACGA-3ʹ, R: 5ʹ-GTCTTACTCCTTGGAGGCC-3ʹ.
Statistical analysisAll MR statistical analyses were performed using the R software (version 4.2.0). MR analysis was performed using the TwosampleMR (version 0.5.7) and MR-PRESSO (version 3.6.0) R packages. The code used to perform all the analysis steps can be found in the online GitHub repository (https://github.com/jmzeng1314/GEO). The receiver operating characteristic (ROC) analysis of the data was performed using pROC [1.18.0], and the results were visualized with ggplot2 [3.3.6]. Dunnett’s test in Ordinary one-way ANOVA to compare differences between multiple groups.
留言 (0)