
記住我





The investigation into the prevalence of co-existing blaKPC variants within clinical samples involved the collection of CRKP strains carrying different blaKPC-variants, transiting from blaKPC−2 to blaKPC−33, from lung transplantation patients subjected to CAZ/AVI treatment between Sep 2021 and Apr 2023. Demographic variables, including gender, age, and antibiotic administration history, were retrospectively extracted from medical reports.
K. pneumoniae strains were identified by Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS, Bruker Daltonics, Germany). AST was performed using the Vitek-2 system with N335 susceptibility cards for meropenem, ceftazidime, aztreonam, and imipenem. MICs of CAZ/AVI were determined using the microdilution broth method (bio-KONT, Ltd. China), with interpretation based on CLSI-recommended [13]. The presence of blaKPC was verified using the GeneXpert system, and positive cases underwent blaKPC genotyping through polymerase chain reaction (PCR) and Sanger sequencing with specific primers: F-CGGTGGTGGGCCAATAGATG and R-TCGTCAACCAGACCCGTTTT.
Whole-genome sequencing (WGS) and bioinformatics analysisGenomic DNA extraction from strains with shifting blaKPC variants was performed using the QIAamp DNA mini kit from Qiagen. Whole-genome sequences were obtained through Illumina NovaSeq PE150 and nanopore sequencing on MinION flow cells. Filtering removed low-quality sequences and adaptors. The online CGE tool (https://cge.cbs.dtu.dk) was employed for multilocus sequence typing (MLST) analysis and identification of acquired antimicrobial resistance genes, while BLASTn against the NCBI database was used for homologous comparisons.
Gene prediction was performed using Prokka 1.13. Antimicrobial resistance genes, MLST, and species identification were analyzed using Kleborate v2.0.4. Furthermore, Snippy 3.2-dev, Gubbins v2.4.1, SNP-sites v2.5.1, RAxMLv8.2.12, and iTOL v5.6 were employed for phylogenetic analysis for high-quality SNPs, filtering recombination events, core polymorphic sites, creating a RAxML tree under the GTR-GAMMA model, and visualization, respectively.
Pulsed-field gel electrophoresis (PFGE) analysisPFGE was conducted to evaluate the clonal relationships among K. pneumoniae strains from Patient 10. Total DNA preparation and PFGE followed established protocols [14]. Strains with a genetic similarity of ≥ 85% or ≤ 4 fragment differences in PFGE profiles were classified as the same clone.
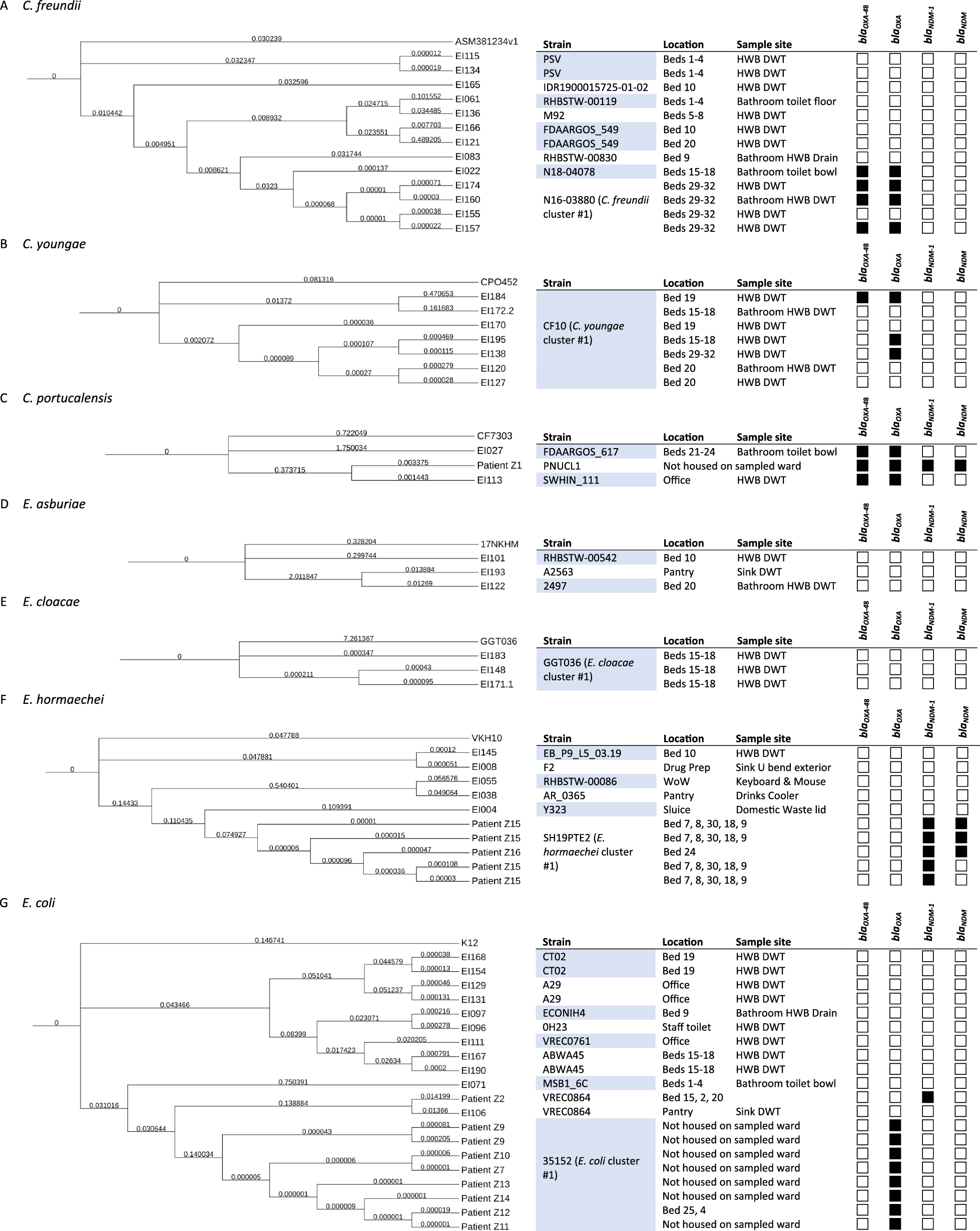
Fig. 1
Strain isolation and antibiotic administration. (A) Clinical data for 10 patients in chronological order. ○: First non-blaKPC−2-carrying CRKP strains for further analysis. (B) Medical history and antibiotic stewardship of Patient 10 and the corresponding strains. (C) Phylogenetic tree and heat map of demographic data of 10 patients, and the molecular characteristics and β-lactamase genes of their corresponding CRKP strains. Notes: CRKP: Carbapenem-resistant Klebsiella pneumoniae
Limiting dilution methodThe limiting dilution method was used to isolate individual clones of CRKP, each carrying either blaKPC−2 or blaKPC−33, as illustrated in Fig. 2.
Briefly, the solution of KP40034-mix strains was prepared to a concentration of 0.5 McFarland, and 110 µl were transferred into the well A1 of a 96-well plate. Simultaneously, except for A1, other 95 wells were added with 100 µl of LB broth. Afterward, 10 µl of KP40034-mix solution was transferred from the preceding well into 100 µl of broth in the subsequent well by using a pipette, from well A1 to well H1, respectively, ensuring thorough mixing. Then, 10 µl solution was transferred from the first column (A1-H1) was transferred into the adjacent column (A2-H2) using an 8-channel pipette. Repeat this process until the last column (A12-H12) was reached, with thorough mixing at each step. After an overnight incubation at 37 °C, OD600 absorbance was measured to confirm bacterial growth. Wells showing growth would undergo genomic DNA extraction and Sanger sequencing of the blaKPC gene to validate monoclonality. Monoclonal strains carrying blaKPC were then sub-cultured on LB agar medium for further analysis.
Fig. 2
Limiting dilution and sequencing of mixed strain Isolate blaKPC-carrying monoclonal strains using limiting dilution. Serial 10-fold dilutions in 96-well plates. Select tested strains from the middle zone. Streak them onto LB agar. Select single colonies after overnight incubation at 37℃. Single colonies and mixed KP40034 strains were sequenced to distinguish blaKPC genes. Sanger sequencing of the 523–541 bp region shows 4 colors, Showing the co-existence of A and C at the N site
Drop-plate experimentTo validate the co-existence of mixed strains carrying either blaKPC−2 or blaKPC−33, we designed a drop-plate experiment, adapted from the agar dilution method but with slight modifications [15]. In brief, five CRKP strains, isolated from Patient 10 using the above limiting dilution method, were selected for further analysis, including one blaKPC−2-harboring strain, two blaKPC−33-harboring ones, and two mixed strains. The McFarland turbidity of each strain was adjusted to 0.5 with 0.45% saline, and serial 10-fold dilutions were prepared for theoretical concentrations ranging from 1 ~ 1.5 × 108 to 104 CFU/ml. A 4-µl fully mixed bacterial suspension was spotted onto designated points on six LB agar medium plates supplemented with different CAZ/AVI levels, as depicted in Fig. 3A. After storage at room temperature for 30 min, the plates were incubated at 37℃ overnight to observe bacterial droplet growth.
Fig. 3
Drop plate experiments with different dilutions. (A) Drop plate experiments: Overnight-grown strains adjusted to 0.5 McFarland turbidity and serially diluted 10-fold. Drops of bacterial suspension were added to 6 LB agar plates with varying CAZ/AVI concentrations at marked positions. (B) Results: blaKPC−33-containing strains formed round colonies, while mixed strains showed spot-like morphology
Following CAZ/AVI breakpoints for a dosage regimen of 2.5 g every 8 h (4:1) administered over 2 h, MICs ≤ 8/4 mg/L and ≥ 16/4 mg/L were defined as susceptible and resistant, respectively [16]. Additionally, we calculated the AUC (0-τ) values of ceftazidime and avibactam as 289.0 and 42.1 µg·h/mL, respectively, by using the pharmacokinetic formula [17]. The average therapeutic concentrations of ceftazidime and avibactam were 36 and 5 µg/mL, respectively, with a ratio of 7.2 to 1. Therefore, we set the CAZ/AVI levels in LB plates to reflect the actual treatment scenario of samples isolated from the respiratory tract: 0, 8/4, 16/4, 32/4, 48/4, and 72/4 mg/L, respectively.
Growth assay and in vitro induction experimentTo emulate the in vivo phenotypic transition from CAZ/AVI-sensitive to resistant states via the mutation of blaKPC−2 to blaKPC−33 under antimicrobial pressure, an in vitro CAZ/AVI induction resistance experiment was conducted over four days, by using strains KP40034-2 S (solely harboring blaKPC−2 and sensitive to CAZ/AVI) and KP40034-mix (containing both blaKPC−2-carrying strains and blaKPC−33-carrying strains), as illustrated in Fig. 4A.
Fig. 4
In vitro induction experiment. (A) Procedure: LB broth with different CAZ/AVI concentrations (36/5 mg/L, 48/6.7 mg/L, 60/8.3 mg/L, and 72/10 mg/L, respectively, the same below) divided into two groups. After overnight induction, suspensions were measured for absorbance and sequenced. (B) Results: (1) OD600 of KP40034-mix with CAZ/AVI at differet concentrations: Significant decrease in strains as CAZ/AVI concentration exceeds MIC of mix strains. (2) OD600 of KP40034-2 S with CAZ/AVI at differet concentrations: Strain count remains low due to susceptibility to CAZ/AVI. (3) Proportion of blaKPC−33 in KP40034-mix at different CAZ/AVI concentrations: Increases with induction time; insufficient CAZ/AVI doses decrease blaKPC−2 but increase blaKPC−33. (4) AST of KP40034-mix: Susceptibility to CAZ/AVI increases over time. Notes: AST: antimicrobial susceptibility testing
The KP40034-2 S and KP40034-mix strains were incubated overnight at 37 °C in 5 mL LB broth. Subsequently, a 50-µL aliquot from each culture, adjusted to 0.5 McFarland turbidity, was transferred into another tube containing 5 mL fresh LB broth with varying CAZ/AVI concentrations. CAZ/AVI levels and ratios were set at 36/4 mg/L, 48/4 mg/L, 60/4 mg/L, and 72/4 mg/L, respectively, in accordance with a previous study [17].
The experiment was conducted over 4 days. On the first day, the tubes were inoculated with KP40034-mix and KP40034-2 S cultures. For the following three days, 50 µL of the culture from the previous day was utilized to inoculate the freshly prepared LB broth containing CAZ/AVI at different concentrations [18].
After that, a 20-µL aliquot was pipetted for qPCR to ascertain if there was the co-existence of blaKPC−2 to blaKPC−33. A 100-µL aliquot, adjusted to 0.5 McFarland turbidity, underwent a 107-fold dilution on LB agar medium. Single colonies were selected after overnight culture for Sanger sequencing to detect the 532nd position mutation of the blaKPC−2 gene. Additionally, MIC results of CAZ/AVI for the induced strains were determined.
Real-time quantitative PCR (RT-qPCR) probe for bla KPC−2 and bla KPC−33To detect the co-existence of blaKPC−2 and blaKPC−33 in mixed strains, we developed two qPCR probes targeting the 532nd bp position of the blaKPC−2 gene. Relative quantification was performed using the 2−ΔCt method, as previously described [19]. Sequences of blaKPC−2 and blaKPC−33 genes from K. pneumoniae were obtained from GenBank (CP133867.1 and NG_056170.1). Primer Express software was utilized to design Allele-Refraction Mutation System Polymerase Chain Reaction (ARMS-PCR) primers and consensus probes, which included a mismatch at position − 3 from the 3’ end to enhance specificity. Locked nucleic acid (LNA) was incorporated, and ARMS-PCR primers had a higher Tm than the forward primers (Supplemental Table 1; LNA denoted by + N). PCR amplification was carried out in two self-contained 25 µL reactions (tube A for blaKPC−2 and tube B for blaKPC−33) with dNTPs, buffer, MgCl2, Reverse consensus primer, Taqman consensus probe, and Taq DNA polymerase. Thermal conditions included 1 cycle at 95 °C for 5 min, followed by 50 cycles at 95 °C for 15 s and 60 °C for 35 s (fluorescence collection).
The probes effectively detected targeted genes in mixed strains, even after a 109-fold dilution from the 0.5 McFarland turbidity level, ensuring separate detection of both blaKPC−2 and blaKPC−33 genes, even at a ratio as high as 105 to 1.
留言 (0)