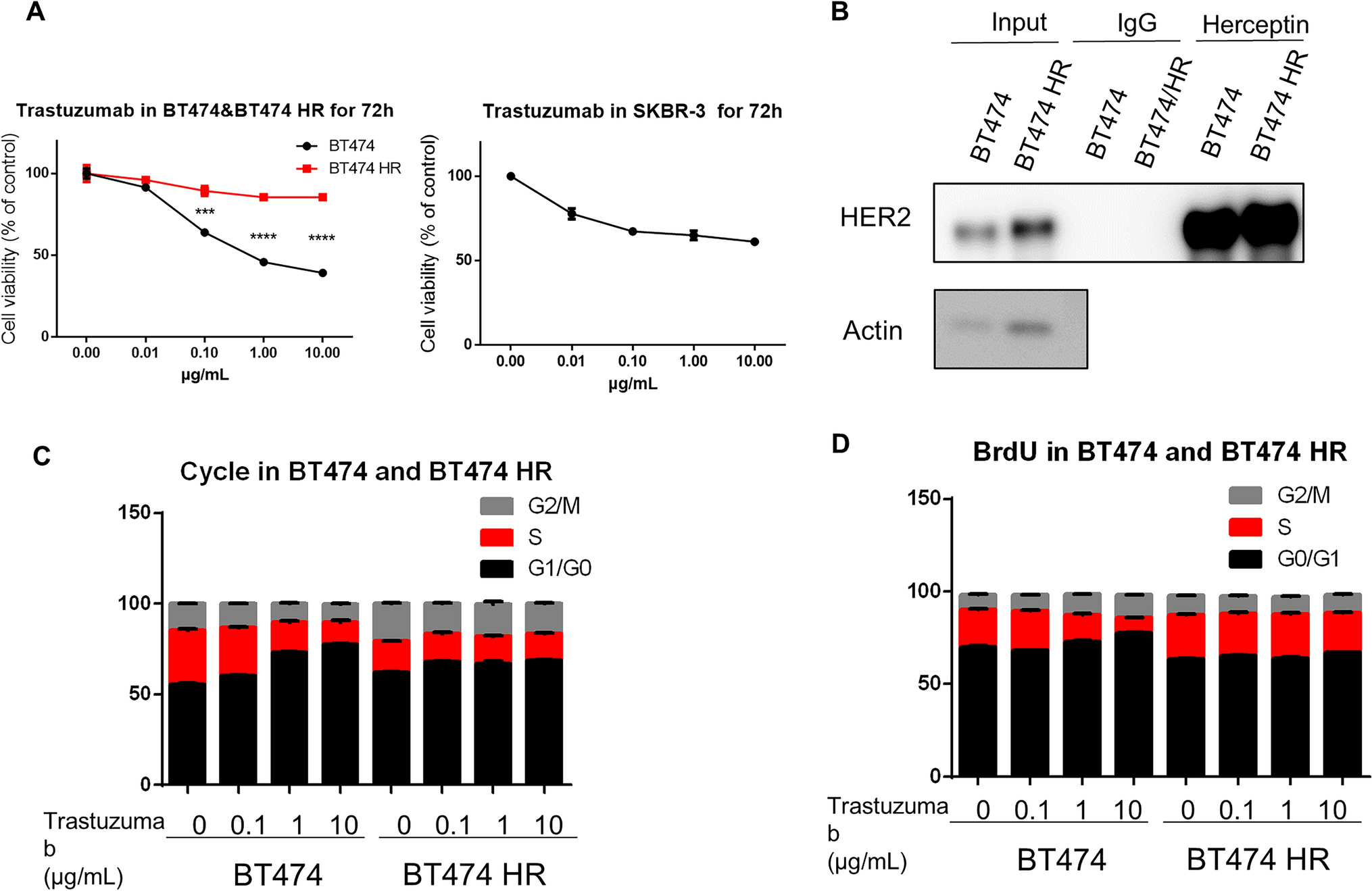
Cell lines and reagents
The human ESCC cell lines, namely KYSE30, KYSE150, TE-1, KYSE450, KYSE410, and KYSE510, were acquired from the Key Laboratory of Shandong Cancer Hospital and Institute. To create an optimal growth environment, the cells were cultured in DMEM with a high glucose concentration, supplemented with 10% foetal bovine serum and 1% penicillin-streptomycin (Gibco Laboratories; Biosharp). The cells were then maintained at a temperature of 37 °C in a humidified incubator with a 5% CO2 atmosphere.
ML385 (CAS: 846557-71-9, lot # 24435, purity: 99.55%) was obtained from MedChemExpress (New Jersey, NJ, USA). It was dissolved in DMSO purchased from Beijing Solarbio Science & Technology (Beijing, China) to prepare a stock solution with a concentration of 20 mM, it was stored at −80 °C for future use. To prepare ML385 for use, it was diluted in the complete cell culture medium to the desired concentrations as indicated. Primary antibodies against NRF2 (16396-1-AP), SLC7A11 (26864-1-AP), and Lamin B1 (12987-1-AP) were purchased from Protein Tech Group Inc. (Chicago, IL, USA). Additionally, the GPX4 antibody (ab125066) was procured from Abcam (Cambridge, MA, USA). Other antibodies against β-actin (#4970), anti-rabbit IgG (#7074), and anti-rabbit IgG (H + L), F(ab’)2 Fragment (#4413) were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Cell viability and colony formation assay
The impact of ML385 on the viability of the KYSE150 and KYSE510 cell lines was evaluated using the Enhanced Cell Counting Kit-8 (CCK-8; Bioss, Beijing, China). In brief, cells were seeded in triplicate at a density of 2000 cells per well in 96-well plates and then incubated at 37 °C with 5% CO2 for a period of 24 h. Next, the cells were exposed to ML385 (1, 2, 5, 10, and 20 µM) or DMSO for 48 h. Thereafter, a volume of 10 µL of CCK-8 solution was carefully added to each well of the plate and incubated at 37 °C for 2 h. Subsequently, the optical density (OD) was assessed at a wavelength of 450 nm using a microplate reader (SpectraMax; Molecular Devices, California, USA). To eliminate the influence of the medium, a blank background group consisting solely of DMEM was employed to subtract the OD value. Next, the inhibition rate of cells was determined using the following formula:
$$}\left( \% \right) = \left( } - }} \right)/\left( } - }} \right)} \times }00$$
Regarding the colony formation assay, the attached cells (600 cells/well) were incubated with a fresh medium containing an appropriate amount of ML385, and the plates were irradiated in a cabinet irradiator (Rad Source Technologies, Georgia, USA) at 0, 2, 4, 6, 8 Gy after 24 h. The six-wells plates were carefully maintained and cultured for 7–14 days, allowing the colonies to grow and reach an optimal state. After discarding the medium and fixing the cells with methanol, the colonies were stained with 0.5% crystal violet for a duration of 10 min. Afterward, they were thoroughly washed with water to remove any excess staining. Lastly, the surviving colonies (over 50 cells) were counted. A triplicate of each experiment was performed. The cell survival curves were analyzed and fitted using the linear-quadratic model in GraphPad Prism 8 software (GraphPad Software, Inc., La Jolla, CA, USA).
Apoptosis and cell cycle
The Annexin V-FITC/propidium iodide (PI) Apoptosis Detection Kit from BD Biosciences was used to detect apoptosis. In a nutshell, the cultured cells were treated with ML385 or IR (8 Gy) alone or ML385 for 24 h followed by IR (8 Gy). After 24 h of IR treatment, cells were gathered and washed twice with phosphate-buffered saline (PBS) to remove any residual media or contaminants, then meticulously suspended in Annexin V binding buffer, incubated with 50 µL/mL FITC-Annexin V and 50 µL/mL PI for 15 min in the dark environment, and assessed using flow cytometer (FACSCalibur; BD Biosciences, New Jersey, USA) and the FlowJo V10 software.
To analyse cell cycle distribution, the treated cells were collected, rinsed twice, then fixed in 70% ethyl alcohol for a duration of 24 h. Subsequently, the cells were washed and resuspended in 0.5 mL PI/RNase staining buffer. After incubation at 23 °C for 15 min in the absence of light, the cells were analysed using a flow cytometer. Additionally, cell cycle profiles were analysed using the ModFit software.
Immunofluorescence
A total of 5.0 × 104 cells were cultured in 12-well plates and subjected to ML385 treatment. After 24 h of IR, the cells were washed and fixed with 4% paraformaldehyde for a duration of 20 min. Subsequently, the washed cells were blocked with 5% bovine serum albumin for a period of 30 min. Furthermore, the cells were subjected to immunodetection, which involved incubation with polyclonal anti-Nrf2 antibody (1:300) overnight at 4 °C and FITC-conjugated secondary anti-rabbit IgG antibody (1:1000) for 1 h on the subsequent day. Lastly, the washed cells were incubated with 200 µL DAPI for 5 min to effectively stain the nuclei. Fluorescence images were acquired using a fluorescence microscope.
Quantitative polymerase chain reaction (qPCR)
The Total RNA Kit (TIANGEN, Beijing, China) was used for the isolation of total RNA. The PrimeScriptTM RT Master Mix (Takara Bio Inc., Shiga, Japan) was used to reverse transcribe 500 ng of total RNA into cDNA. Following amplification, the obtained cDNA underwent qPCR analysis using TB Green Premix Ex Taq II (Takara Bio Inc., Shiga, Japan).
The primer sequences for NRF2 and β-actin were as follows: NRF2 forward, 5′-TCCAGTCAGAAACCAGTGGAT-3′ and reverse 5′-GAATGTCTGCGCCAAAAGCTG-3′; β-actin forward, 5′-CGTGGACATCCGCAAAGAC-3′ and reverse 5′-CGTCATACTCCTGCTTGCTG-3′.
Western blotting
The washed cells were lysed in sodium dodecyl sulfate (SDS) lysis buffer supplemented with the protease inhibitor phenylmethylsulfonyl fluoride (Beyotime Biotechnology, Nanjing, China). Next, a cytoplasmic and nuclear extraction kit was employed to isolate nuclear proteins. The adjusted concentration of protein extracts were loaded onto SDS-polyacrylamide gel electrophoresis membranes and transferred to polyvinylidene fluoride (PVDF) membranes. Subsequently, the PVDF membranes were incubated with primary antibody overnight at 4 °C after blocking with 5% non-fat milk powder in TBS-Tween 20 (TBST) for 1 h, washed thrice with TBST, and reincubated with horseradish peroxidase-conjugated IgG for 1 h at ambient temperature. Lastly, the bands were visualised using an ECL detection reagent (Millipore, USA).
ROS and lipid peroxidation
The cultured cells were treated with ML385 or IR (8 Gy) alone or ML385 for 24 h followed by IR (8 Gy). Next, 10 µM CM-H2DCFDA (ThermoFisher, C6827, USA) or 5 µM BODIPY 581/591 C11 dye (Invitrogen, D3861, USA) was added to the dishes containing fresh medium to detect total ROS or lipid peroxidation levels, respectively. After 30 min incubation, the washed cells were analysed via flow cytometry.
GSH assay
The cells adhering to the 96-well plate were treated with ML385, IR, or ML385 + IR separately. The treated cells were cultured in fresh medium containing 100 µL of prepared 1× GSH-Glo Reagent (Promega) at room temperature for a period of 30 min, and then 100 µL of reconstituted Luciferin Detection Reagent (Promega) was added to each well for another 15 min. Lastly, luminescence was measured and normalised to the cell viability. The relative GSH levels in cells treated with the test compounds were normalised to those in control cells.
Animal experiments
The athymic nude mice used in this study were obtained from Charles River (Beijing, China), and 2 × 106 KYSE150 cells were injected subcutaneously into 6-week-old specific pathogen-free BALB/c nude mice. The tumours reached a size of approximately 50 mm3, the mice were randomly divided into four groups and intraperitoneally treated with saline or ML385 (30 mg/kg) for two consecutive days. After 24 h, the mice were anaesthetised by intraperitoneal injection of 0.1 mL 1% sodium pentobarbital. After the mice became stationary, their heads and limbs were fixed with a special mouse position fixator, and the tumours were exposed to the centre of the irradiation field, shielding the other parts with lead blocks. Irradiation was performed using an animal cell irradiator (model: Rad Source RS2000Pro, parameter setting: 225.0 KV, 17.7 mA, dose rate: 2.4 Gy/min, and total dose: 8 Gy). The tumour volume was calculated as follows:
$$} = } \times }^}} \times }/}$$
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 8. Each experiment was repeated at least three times. The results were presented as the mean ± standard deviation (SD), and error bars indicate the standard deviation. The statistical significance of the results was determined using the analysis of variance(ANOVA). Differences between groups were considered statistically significant when the p-value was less than 0.05 (p < 0.05).




留言 (0)