
記住我
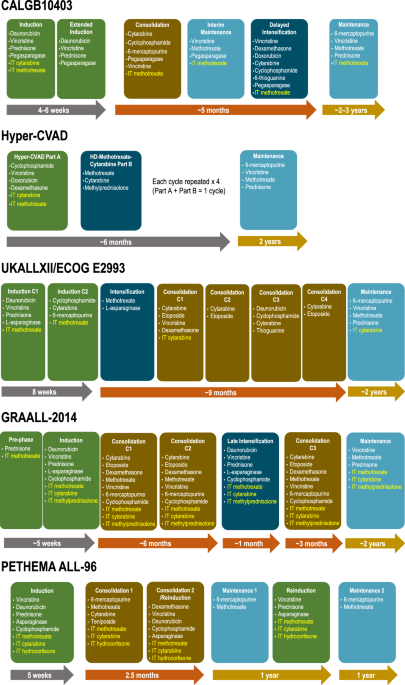



ACHM-025 (((2-((4-methylpiperazin-1-yl)sulfonyl)-4-nitrophenyl)azanediyl)bis(ethane-2,1-diyl) dimethanesulfonate) is a novel prodrug selectively activated by AKR1C3 in the presence of NADPH to form the nitrogen mustard DNA alkylating agent, ACHM-025H (Fig. 1A). The nitrogen mustard, ACHM-025H, is able to bis-alkylate DNA via the amino group of two guanine bases, forming DNA intra- or inter-strand crosslinks (Fig. 1A). CP is a DNA alkylating prodrug, currently used in the treatment of pediatric ALL (Supplementary Fig. S1). Unlike ACHM-025, CP is activated by liver enzymes in a multi-step process that culminates in fragmentation to form the nitrogen mustard DNA alkylating agent, phosphoramide mustard (Supplementary Fig. S1) [24]. Importantly, and unlike earlier AKR1C3-activated prodrugs such as OBI-3424 [11] and PR-104 [12, 13, 25], ACHM-025 forms the active agent ACHM-025H in a single step (Supplementary Figs. S1 and S2).
Fig. 1: Ex vivo activity and selectivity of ACHM-025.
A Mechanism of activation of prodrug ACHM-025: the nitro group of ACHM-025 is reduced by AKR1C3 in the presence of NADPH to a hydroxylamine group, forming the active agent ACHM-025H in a single step (nitro group highlighted blue and hydroxylamine group highlighted red). ACHM-025H is a nitrogen mustard, which is able to bis-alkylate DNA (nitrogen mustard highlighted red). B–D A panel of 6 T-ALL PDXs (red) and 6 B-ALL PDXs (gray) were treated ex vivo with ACHM-025± an AKR1C3 specific inhibitor (AKR1C3i). B ACHM-025 IC50 values plotted against AKR1C3 protein expression as measured by immunoblotting. C ACHM-025 IC50 values ± AKR1C3i against B-ALL (gray) and T-ALL (red) PDXs. D Fold-increase in IC50 (ACHM-025 + AKR1C3i/ACHM-025 alone) versus AKR1C3 protein expression. E, F HCT116 parental (WT) and HCT116 cells overexpressing the AKR1C family members, AKR1C1-AKR1C4, were treated in vitro with ACHM-025 ± an AKR1C3 specific inhibitor (AKR1C3i). E ACHM-025 IC50 values. F Fold-increase in IC50 (ACHM-025 + AKR1C3i/ACHM-025 alone) of HCT116 WT and HCT116 overexpressing AKR1C3. B–F Each data point represents the mean of at least three independent experiments. p-values were corrected for multiple comparisons. B, D Centre and curved lines represent linear regression and 95% confidence interval, respectively.
To evaluate the activity of ACHM-025 compared with OBI-3424 and PR-104A, ex vivo cytotoxicity assays were carried out on 12 PDXs (6 T-ALL PDXs and 6 B-ALL PDXs) with varying levels of AKR1C3 protein expression. The IC50 values for ACHM-025, OBI-3424 and PR-104A all displayed a significant inverse correlation with basal AKR1C3 protein expression (Fig. 1B, Supplementary Fig. S3). The strongest inverse correlation was observed with PR-104A (R2 = 0.8166; p < 0.0004; Supplementary Fig. S3A), followed by ACHM-025 (R2 = 0.7630; p = 0.0008; Fig. 1B), then OBI-3424 (R2 = 0.4834; p = 0.048; Supplementary Fig. S3B). However, PR-104A showed a modest interdependence (slope) and reduced overall dose potency, where the median IC50 values were 26.6 µM for B-ALL and 1.8 µM for T-ALL PDXs (Supplementary Table S2). Comparatively, the median IC50 values for ACHM-025 were 840 nM for B-ALL and 29 nM for T-ALL. The median IC50 values for OBI-3424 were 60 nM for B-ALL and 8 nM for T-ALL, although the overall correlation with AKR1C3 expression was weak (Supplementary Table S2).
To further assess the selectivity of ACHM-025 for AKR1C3, all 12 PDXs were pre-treated ex vivo with the isoform-specific AKR1C3 inhibitor, SN34037, prior to treatment with ACHM-025 (Supplementary Fig. S4) [16]. Inhibition of AKR1C3 enzymatic activity prevented ACHM-025 cytotoxicity with a significant increase in IC50 in both B-ALL and T-ALL PDXs (Fig. 1C, Supplementary Table S2). Furthermore, the fold-increase in IC50 (ACHM-025 + SN34037/ACHM-025 alone) showed a strong and significant correlation with basal AKR1C3 protein expression (R2 = 0.8171; p < 0.0004; Fig. 1D). Further, ACHM-025 showed selectivity for activation by the AKR1C3 isoform, compared to AKR1C1, AKR1C2 and AKR1C4 in HCT116 cells (Fig. 1E, Supplementary Table S3). Incubation of HCT116/AKR1C3 overexpressing cells with SN34037 caused a 1,700-fold increase in IC50 (Fig. 1F, Supplementary Table S3). Moreover, ACHM-025 ex vivo cytotoxicity showed a significant inverse correlation with AKR1C3 mRNA expression (R2 = 0.8720; p < 0.0004; Supplementary Fig. S5A), compared to isoforms AKR1C1, AKR1C2 and AKR1C4 (Supplementary Fig. S5B–D). Taken together, these data show that ACHM-025 has improved specificity for AKR1C3 and dose potency compared to OBI-3424 and PR-104A, respectively.
ACHM-025 displays profound single agent in vivo efficacy against chemoresistant T-ALL PDXsWe next sought to determine the in vivo efficacy of ACHM-025 as a single agent. Initial in vivo pharmacokinetic analysis of ACHM-025 in murine plasma and brain tissue was evaluated at 3 dose levels (5, 10, 25 mg/kg) and 6 time points (0.25–2 h) via intraperitoneal injection (IP). ACHM-025 was cleared rapidly from the plasma, with Cmax ranging 1.6–9.3 µM across the dose levels tested, with a total plasma exposure (AUC0-inf) ranging 0.5–3.3 µmol-h/L (Supplementary Fig. S6A, Supplementary Table S4). ACHM-025 concentration in brain tissue was low, although the peak concentration (0.14 µM, 0.25 h, 25 mg/kg ACHM-025) exceeded the IC50 values across all T-ALLs (median 0.029 µM; Supplementary Tables S2 and S4). ACHM-025 displayed limited protein binding, with 43.1% and 35.4% bound to mouse and human plasma proteins, respectively. The maximum dose of ACHM-025 evaluated (25 mg/kg via IP weekly ×3) was well tolerated in naïve immunodeficient mice including no major hematology effects (Supplementary Fig. S6B, Supplementary Tables S5, S6) [26]. Therefore, the IP dose of 25 mg/kg was selected as the maximum dose to assess the in vivo efficacy of ACHM-025 in murine PDX models.
The in vivo efficacy of ACHM-025 (25 mg/kg, IP Days 0, 7, 14) was first evaluated against the aggressive and chemoresistant T-ALL PDX, ALL-31, which has high AKR1C3 expression (Supplementary Table S7). ACHM-025 caused a rapid reduction of human leukemia cells in the murine peripheral blood (PB) to <1%, which was maintained for 10 consecutive weeks with a concomitant significant increase in event-free survival (EFS) compared to vehicle control treated mice (vehicle median EFS = 16.7 days vs. ACHM-025 median EFS > 332 days, p = 0.0014) (Fig. 2A, Supplementary Table S8). Remarkably, 3 out of 5 mice treated with ACHM-025 did not relapse over the >300-day observation period following the last treatment. When these mice were humanely killed on day 332, flow cytometry analysis revealed no detectable human leukemia cells in the PB, bone marrow (BM), spleen, kidney or liver (data not shown). Three sentinel mice included in the study for Day 28 time point analyses also revealed no detectable human leukemia cells in 3 out of 3 mice in the PB, BM or spleen, which was 14 days after the last ACHM-025 treatment when analyzed as either the % human CD45+ cells versus total mouse cells (Fig. 2B) or the % human CD45+ cells versus mouse CD45+ cells (Supplementary Fig. S7A).
Fig. 2: ACHM-025 displays profound single agent efficacy against chemoresistant T-ALL PDXs in vivo.
A Mice engrafted with a diagnosis/refractory T-ALL PDX (ALL-31) were treated in vivo with ACHM-025 (red; 25 mg/kg, IP Days 0, 7, 14) or vehicle (black). Engraftment of individual mice for each treatment, showing the % human CD45+ over time (left). Mouse EFS, where dashes denote censored mice that did not reach event (right). B Engraftment (% human CD45+ cells versus all mouse cells) of ALL-31 in PB, BM and spleen at Day 0 (open circles), vehicle treated at event (black circles), 25 mg/kg ACHM-025 treated at Day 28 (red circles). C Mice engrafted with a relapse/refractory T-ALL PDX (ALL-8) were treated in vivo with 25 mg/kg ACHM-025 (red; IP Days 0, 7, 14), 15 mg/kg ACHM-025 (orange; IP Days 0, 7, 14), 10 mg/kg ACHM-025 (blue; IP Days 0, 7, 14) or vehicle (black). Engraftment of individual mice for each treatment, showing the % human CD45+ over time (left). Mouse EFS, where dashes denote censored mice that did not reach event (right). D Engraftment (% human CD45+ cells versus all mouse cells) of ALL-8 in PB, BM and organs at Day 0 (open circles), vehicle treated at event (black circles), 10 mg/kg ACHM-025 treated at Day 28 (blue circles), 15 mg/kg ACHM-025 treated at Day 28 (orange circles), and 25 mg/kg ACHM-025 treated at Day 28 (red circles). E, F Engraftment of ALL-8 in the skull demonstrated with routine H&E staining (upper panels and lower left panels) and nuclear TdT immunohistochemical staining (lower right panels). E Untreated mouse at Day 0. F Vehicle treated mouse at event (Day 14). G Mouse treated with 10 mg/kg ACHM-025 (IP Days 0, 7, 14) at Day 28. Widespread leukemic infiltration of the calvarial bone marrow is present in the untreated (E) and vehicle treated (F) mice; in contrast, leukemic infiltration is absent in the 10 mg/kg ACHM-025 treated mouse at Day 28 (G). B, D Gray bars represent the median of 3 mice. Statistical differences between vehicle treated mice at event and ACHM-025 treated mice at Day 28 were determined using unpaired t-tests with Welch’s correction (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Given the remarkable single agent efficacy of ACHM-025, we next undertook an in vivo dose-response study against an additional aggressive T-ALL PDX (ALL-8) (Supplementary Table S7). All three doses of ACHM-025 (10, 15 or 25 mg/kg, IP Days 0, 7, 14) induced significantly prolonged leukemia regressions (maintained complete responses, MCRs) (Fig. 2C, Supplementary Tables S8, S9) and a clear dose response (Fig. 2C, Supplementary Table S8). Similar to ALL-31, 5 out of 6 mice treated with the highest dose of 25 mg/kg had no detectable human leukemia cells in the PB at 250 days after the last treatment (Fig. 2C). Analysis of mice at Day 28, 14 days after the last treatment there was no detectable human leukemia cells in the PB, BM, spleen, kidney or liver at any of the three doses of ACHM-025 when expressed relative to total (Fig. 2D) or CD45+ (Supplementary Fig. 7B) murine cells.
Since ALL can cause relapse via the central nervous system (CNS), we histopathologically analyzed murine skull and brain samples at Day 0 (pre-treatment), vehicle treated at Day 14 (event), and 10 mg/kg ACHM-025 treated and harvested at Day 28. Hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase (TdT) staining showed that, while leukemic infiltration was observed in the calvarial bone marrow at Day 0 (Fig. 2E) and more extensively at Day 14 of the vehicle treated mice (Fig. 2F), there was no observable leukemic infiltration at Day 28 for ACHM-025 treated mice (Fig. 2G). Therefore, we reasoned that relapse is likely to occur through measurable/minimal residual disease (MRD) cells residing in the BM or elsewhere that were not detected at the level of sensitivity of flow cytometry.
To determine whether disease relapse could be due to acquired drug resistance via down-regulation of AKR1C3, we analyzed AKRIC3 mRNA expression in leukemia cells isolated from the spleens of vehicle treated mice and mice treated with ACHM-025 that subsequently relapsed. There was no significant difference in AKR1C3 expression at the mRNA level suggesting that relapse was not due to outgrowth of low AKR1C3 expressing cells (Supplementary Fig. S7C). Taken together, these data show impressive single agent efficacy of ACHM-025 against aggressive and chemoresistant T-ALL PDXs.
A mouse “clinical” trial to evaluate AKR1C3 expression as a biomarker for in vivo ACHM-025 efficacyWe next evaluated the relationship between AKR1C3 expression and in vivo ACHM-025 activity using a single mouse trial (SMT) format (one vehicle control treated mouse, one drug treated mouse) [22] across an extended panel of 25 T-ALL PDXs that better addresses the impact of genetic heterogeneity in pediatric ALL on drug response (Supplementary Fig. S8A). All 25 PDXs treated with vehicle control reached event (huCD45+ >25% in the PB) between 3.1 and 26 days following treatment initiation (Fig. 3A, Supplementary Table S9, Supplementary Fig. S8B). Of the ACHM-025 treated mice, 7/25 PDXs did not reach event during the 250 day monitoring period following the final treatment (Fig. 3A, Supplementary Table S9, Supplementary Fig. S8B). In 22/25 PDXs there were no human leukemia cells detected in the murine PB for at least two weeks after treatment initiation, and all of these mice achieved CRs or MCRs (Supplementary Fig. S9A, Supplementary Table S9). When all vehicle vs. all treated mice were evaluated, ACHM-025 significantly increased EFS compared to vehicle control mice (p < 0.0001; Fig. 3B). Moreover, the 3 PDXs in which only Progressive Disease 1 (PD1) was achieved all expressed minimal levels of AKR1C3 mRNA and protein (Supplementary Fig. S9B–D, Supplementary Table S9).
Fig. 3: A single mouse trial across a panel of T-ALL PDXs identified the levels of AKR1C3 expression as a biomarker for in vivo ACHM-025 efficacy.
A Event free survival (EFS) for 25 T-ALL PDXs treated in vivo with vehicle (black bars) or ACHM-025 (red bars; 25 mg/kg, IP Days 0, 7, 14). Red crosses denote PDXs that did not reach event, black crosses denote non-leukemia related events (NLRE). PDXs are listed in order of increasing AKR1C3 protein expression; low (blue, n = 4), medium (orange, n = 10) or high (purple, n = 11). B Survival of 25 T-ALL PDXs treated in vivo with vehicle (black) or ACHM-025 (red; 25 mg/kg, IP Days 0, 7, 14). C Objective response measures versus AKR1C3 protein expression; high (purple, n = 11), medium (orange, n = 10) or low (blue, n = 4). D Leukemia growth delay (Treated EFS/Vehicle EFS) of PDXs with high (purple, n = 11), medium (orange, n = 10) or low (blue, n = 4) AKR1C3 protein expression. E Basal AKR1C3 intracellular expression as measured by flow cytometry for 25 T-ALL PDXs. Histograms for AKR1C3 unconjugated primary antibody (blue) and APC-conjugated secondary antibody (red). PDXs are listed in order of decreasing AKR1C3 intracellular expression; high (green, n = 20) or low (gray, n = 5). F Objective response measures versus AKR1C3 intracellular expression according to relative fluorescent intensity (RFI) cut-off at 14 as measured in E; high (green, n = 20) or low (gray, n = 5). G Leukemia growth delay (Treated EFS / Vehicle EFS) of PDXs with high (green, n = 20) or low (gray, n = 5) AKR1C3 intracellular expression. B, D, G Significance calculated by Log-rank (Mantel-Cox). Dashes denote censored PDXs that did not reach event or are NLREs.
Comparing the panel of 25 T-ALL PDXs that achieved an objective response (CR or MCR) and those that did not (PD1) with AKR1C3 protein expression, ACHM-025 was significantly less effective against T-ALL PDXs with low AKR1C3 expression compared to those with medium (p = 0.0028) or high (p < 0.0001) AKR1C3 expression (Fig. 3C, D). This was also found using intracellular flow cytometry for measuring AKR1C3 expression with ACHM-025 significantly less effective against T-ALL PDXs below minimum AKR1C3 intracellular expression values required for an objective response (CR or MCR) (p < 0.0001; Fig. 3E–G).
Furthermore, a B-ALL PDX (ALL-11) that had been lentivirally transduced to overexpress AKR1C3 (ALL-11/AKR1C3; AKR1C3high) [27] exhibited an ex vivo ACHM-025 IC50 (13.1 nM) that was more reflective of AKR1C3high T-ALL PDXs (median IC50 29 nM) compared with empty vector control cells (ALL-11/EV; AKR1C3low), in which the IC50 (1261 nM) was more consistent with AKR1C3low B-ALL PDXs (median IC50 840 nM) (Supplementary Fig. S10A, B, Supplementary Table S2). Similarly, the AKR1C3 inhibitor, SN34037, caused >20-fold shift in the ACHM-025 IC50 value in ALL-11/AKR1C3 cells relative to ALL-11/EV cells (Supplementary Fig. S10B, Supplementary Table S2), while ACHM-025 was profoundly more effective in vivo against ALL-11/AKR1C3 than ALL-11/EV (EFS 104 and 49.1 days, respectively; Supplementary Fig. S10C, Supplementary Table S9). Taken together, these findings support AKR1C3 expression as a biomarker for ACHM-025 activity both in vitro and in vivo, and suggest that the potency of ACHM-025 is dependent on AKR1C3 expression levels.
ACHM-025 significantly improves standard-of-care consolidation therapy compared to cyclophosphamideBoth ACHM-025 and CP are DNA bis-alkylating prodrugs, with CP included in standard-of-care consolidation therapy for ALL in combination with, amongst other drugs, cytarabine (Ara-C) and 6-mercaptopurine (6MP). Therefore, we compared the combination in vivo efficacy of ACHM-025/Ara-C/6MP with CP/Ara-C/6MP against ALL-8. ACHM-025 (5 mg/kg, IP Days 0, 7) or CP (75 mg/kg, IP Days 0, 7) combined with Ara-C (12.5 mg/kg, IP Days 0–4, 7–11) and 6MP (12.5 mg/kg, IP Days 0–4, 7–11). Combination dosing and single agents used as twice the dose were well tolerated in naïve immunodeficient mice (Supplementary Fig. S11A, Supplementary Tables S10, S11).
With the exception of 6MP, all single agents caused significant delays in the progression of ALL-8, with treated minus control (T-C) median EFS values of 22.5 days for Ara-C (p = 0.0005), 25.3 days for CP (p = 0.0005) and 57.4 days for ACHM-025 (p = 0.0005) (Table 1, Fig. 4A). While ACHM-025 decreased the levels of human leukemia cells in the murine PB to <1% for six consecutive weeks and elicited an MCR, CP was only able to do so for two weeks (CR; Table 1, Fig. 4A, Supplementary Fig. S11B). Flow cytometric analysis of a separate cohort of mice at Day 28 post treatment initiation showed that only ACHM-025 reduced human leukemia levels in the PB, BM and spleen to undetectable levels at Day 28 (Fig. 4B).
Table 1 In vivo activity of ACHM-025 in combination against a relapsed/refractory T-ALL PDX (ALL-8).Fig. 4: ACHM-025 significantly improves standard-of-care consolidation therapy compared to CP.
A Mice engrafted with a relapse/refractory T-ALL PDX (ALL-8) were treated in vivo with vehicle (black), 6-mercaptopurine (dark blue; 12.5 mg/kg, IP Days 0–4, 7–11), Ara-C (yellow; 12.5 mg/kg, IP Days 0–4, 7–11), CP (light blue; 75 mg/kg, IP Days 0, 7), or ACHM-025 (orange; 5 mg/kg, IP Days 0, 7). Engraftment of individual mice for each treatment, showing the % human CD45+ over time (left) and mouse EFS (right). B Engraftment (% human CD45+ cells versus mouse CD45+ cells) of ALL-8 in PB, BM and spleen at Day 0 (open circles), vehicle treated (black circles; event), 6-mercaptopurine (dark blue; event), Ara-C (yellow; Day 28), CP (light blue; Day 28), ACHM-025 (orange; Day 28), the combination of Ara-C, 6-mercaptopurine and CP (purple; Day 28), or the combination of Ara-C, 6-mercaptopurine and ACHM-025 (red; Day 28). Gray bars represent the median of 3 mice. Statistical differences between vehicle treated mice at event and drug treated mice at Day 28 were determined using unpaired t-tests with Welch’s correction (**p < 0.01; ***p < 0.001; ****p < 0.0001). C Mice engrafted with ALL-8 were treated in vivo with vehicle (black), the combination of Ara-C, 6-mercaptopurine and CP (purple) or the combination of Ara-C, 6-mercaptopurine and ACHM-025 (red). Engraftment of individual mice for each treatment, showing the % human CD45+ over time (upper) and mouse EFS (lower). D Minimal residual disease in the bone marrow at Day 28 for ACHM-025 treated mice (orange circles; n = 3) and ACHM-025/Ara-C/6-mercaptopurine treated mice (red circles; n = 3). Samples displayed as the fraction of a highly engrafted (96% huCD45+) murine bone marrow (BM). Technical triplicates of n = 3 mice per treatment, where dotted lines represent the limit of quantification (LoQ), limit of sensitivity (LoS) and MRD-. A, C Significance calculated by Log-rank (Mantel–Cox).
We next compared the 3-drug combinations and found that ACHM-025/Ara-C/6MP induced sustained remissions in the mice (7 consecutive weeks) compared with only two weeks for CP/Ara-C/6MP (Fig. 4C, Supplementary Fig. S11C), resulting in a median EFS that was 38 days longer (p = 0.0005; Table 1, Fig. 4C). Furthermore, ACHM-025/Ara-C/6MP significantly improved the clearance of leukemia cells from the BM compared with CP/Ara-C/6MP, which still had 28% huCD45+ cells in the BM at Day 28 (Fig. 4B). Using clinically validated and patient-specific quantitative-PCR based minimal residual disease (MRD) testing [28, 29], there was a trend for lower MRD levels in the ACHM-025/Ara-C/6MP combination compared to ACHM-025 alone (Fig. 4D), albeit with most values at borderline limits of quantification.
ACHM-025 in combination with nelarabine is highly effective against a chemoresistant T-ALL PDXCurrently, the only chemotherapy FDA and EMA approved for the treatment of relapsed/refractory T-ALL is the nucleoside analog, nelarabine [30]. Therefore we next compared the in vivo efficacy of nelarabine and ACHM-025 as single agents and in combination against ALL-8. Due to the exceptional efficacy of ACHM-025 as a single agent, its dose was lowered to better assess any ACHM-025/nelarabine combinatorial effect. The combination of ACHM-025 (10 mg/kg, IP Days 0, 7, 14) and nelarabine (125 mg/kg, IP Days 0–4, 14–18) was well tolerated in naïve immunodeficient mice (Supplementary Fig. S12A).
As a single agent, nelarabine was able to significantly delay the progression of leukemia in mice engrafted with ALL-8 but only achieved stable disease (SD) (Table 1, Fig. 5A, B). However, nelarabine did not significantly decrease human leukemia levels in the PB, BM or spleen at Day 28 (Fig. 5C, Supplementary Fig. S12B). In contrast, both ACHM-025 and the ACHM-025/nelarabine combination profoundly delayed leukemia progression and induced MCRs, with no mice relapsing in the combination cohort of ACHM-025/nelarabine over the observable period of 300 days following the final day of treatment (Table 1, Fig. 5A, B, Supplementary Fig. S12B). At Day 28 there was no detectable human leukemia cells in the BM of ACHM-025 and ACHM-025/nelarabine treated mice by flow cytometry (Fig. 5C) and although BM samples were MRD positive, they were not within the quantitative range of the PCR-based assay (Fig. 5D). Finally, once all surviving mice treated with the ACHM-025/nelarabine combination were euthanized we found no detectable human leukemia cells in the PB, BM or other organs (spleen, kidney and liver) as measured by flow cytometry (>275 days after the last treatment, data not shown). Taken together, the combination of ACHM-25 and nelarabine appeared to eradicate this chemoresistant T-ALL PDX compared to each single agent alone.
Fig. 5: ACHM-025 in combination with nelarabine is highly effective against chemoresistant T-ALL.
A Mice engrafted with a relapse/refractory T-ALL PDX (ALL-8) were treated in vivo with vehicle (black), nelarabine (blue; 125 mg/kg, IP Days 0–4, 14–18), ACHM-025 (orange; 10 mg/kg, IP Days 0, 7, 14), or the combination ACHM-025/nelarabine (red). Engraftment of individual mice for each treatment, showing the % human CD45+ over time (left). Mouse EFS, where dashes denote censored mice that did not reach event or are NLREs (right). Significance calculated by Log-rank (Mantel–Cox). B Percentage change from baseline of the minimum human CD45+ values in ALL-8 PB engraftment from treatment initiation of individual mice (n = 6 per treatment group). C Engraftment (% human CD45+ cells versus mouse CD45+ cells) of ALL-8 in the PB, BM and spleen at Day 0 (open circles), vehicle treated at event (black circles), nelarabine treated at Day 28 (blue circles), ACHM-025 treated at Day 28 (orange circles), and ACHM-025/nelarabine treated at Day 28 (red circles). Gray bars represent the median of 3 mice. Statistical differences between vehicle treated mice at event and drug treated mice at Day 28 were determined using unpaired t-tests with Welch’s correction (**p < 0.01; ***p < 0.001; ****p < 0.0001). D Minimal residual disease in the bone marrow at Day 28 for ACHM-025 treated (orange circles; n = 3) and ACHM-025/nelarabine treated (red circles; n = 3). Samples displayed as the fraction of a highly engrafted (96% huCD45+) murine bone marrow (BM). Technical triplicates of n = 3 mice per treatment, where dotted lines represent the limit of quantification (LoQ), limit of sensitivity (LoS) and MRD-.
Evaluation of AKR1C3 expression as a biomarker for ACHM-025 efficacy in other pediatric cancersBased on the promising results of the in vivo ACHM-025 SMT study against 25 T-ALL PDXs, we interrogated a recently published large cohort of 1,335 pediatric T-ALL patients to reveal potential subtype-specific AKR1C3 mRNA expression [31]. AKR1C3 expression appeared consistent across most T-ALL subtypes, with notably lower expression in the SPI1, TLX1 and TLX3 subtypes (Supplementary Fig. S13). We next evaluated whether AKR1C3 expression could be used as a biomarker for ACHM-025 efficacy in other pediatric cancers. Similar to Fig. 3, we separated the panel of 25 T-ALL PDXs into those that achieved an objective response (CR or MCR) and those that did not (PD1) following ACHM-025 in vivo treatment and assessed AKR1C3 RNA-seq expression (Fig. 6A). The minimum AKR1C3 RNA-seq expression value required for an objective response was determined as transcripts per million (TPM) > 48, where T-ALL PDXs above this threshold were designated AKR1C3high (Fig. 6A). As we have shown with AKR1C3 protein and intracellular expression, ACHM-025 was significantly more effective in vivo against T-ALL PDXs with high AKR1C3 expression compared to those with low AKR1C3 expression (p < 0.0001; Fig. 6B). Importantly, AKR1C3 RNA-seq expression significantly correlated with ex vivo ACHM-025 efficacy against T-ALL PDXs (Fig. 6C).
Fig. 6: ACHM-025 displays ex vivo activity against multiple pediatric cancers with elevated AKR1C3 expression.
A Objective response measures of 25 T-ALL PDXs treated in vivo with ACHM-025 (25 mg/kg, IP Days 0, 7, 14) versus AKR1C3 mRNA expression as measured by RNA-seq; high (red, n = 22) or low (gray, n = 3). B Leukemia growth delay (Treated EFS / Vehicle EFS) of PDXs with high (red, n = 22) or low (gray, n = 3) AKR1C3 mRNA expression (RNA-seq). Significance calculated by Log-rank (mantel-Cox). Dashes denote censored PDXs that did not reach event or are NLREs. C A panel of 8 T-ALL PDXs with high (red, n = 6) or low (gray, n = 2) AKR1C3 mRNA expression were treated ex vivo with ACHM-025. ACHM-025 IC50 values plotted against AKR1C3 mRNA expression (RNA-seq). Centre and curved lines represent linear regression and 95% confidence interval, respectively. D AKR1C3 mRNA expression of 65 ALL PDXs as measured by RNA-seq; high (red, n = 4) or low (gray, n = 61). The dashed line represents the minimum AKR1C3 expression level to be classified as AKR1C3high (TPM > 48). E ACHM-025 IC50 values of ALL PDXs with high (red, n = 9) or low (gray, n = 7) AKR1C3 mRNA expression (RNA-seq). F AKR1C3 mRNA expression of 371 pediatric patient samples as measured by RNA-seq. Each data point represents AKR1C3 expression of an individual patient sample, where the solid blue line represents the median AKR1C3 expression of each type of cancer. The dashed line represents the minimum AKR1C3 expression level to be classified as AKR1C3high (TPM > 48). G ACHM-025 IC50 values of T-ALL, acute myeloid leukemia (AML), neuroblastoma (NB), Ewing sarcoma (ES), diffuse midline glioma (DMG), hepatoblastoma (HB), and ependymoma (EPD) PDXs. PDXs denoted AKR1C3 high (red, n = 4) or low (gray, n = 11) as measured by qRT-PCR (CNRQ). Each data point represents the mean of three technical repeats. (C, E) Each data point represents the mean of at least three independent experiments.
To evaluate whether AKR1C3 expression could be used as a biomarker for ACHM-025 ex vivo efficacy against non-T-ALL PDXs, we applied the AKR1C3 RNA-seq threshold identified in Fig. 6A (TPM > 48) to a panel of 65 non-T-ALL PDXs (Fig. 6D). Although only 6% of non-T-ALL PDXs (4/65) were assigned AKR1C3high, these PDXs showed similar levels of ACHM-025 ex vivo sensitivity as AKR1C3high T-ALL PDXs (Fig. 6E, Supplementary Figs. S4 and S14, Supplementary Table S12). We next evaluated AKR1C3 RNA-seq expression in 759 patients enrolled in the ZERO Childhood Cancer Program [32], which is a precision medicine program for children with poor-outcome, rare, relapsed or refractory cancer (subset shown in Fig. 6F, whole cohort shown in Supplementary Fig. S15). We applied the AKR1C3 RNA-seq threshold identified in Fig. 6A (TPM > 48) to the 759 patient samples and 11% (87/759) were assigned AKR1C3high (Fig. 6F, Supplementary Fig. S15), with hepatocellular cancers displaying the highest AKR1C3 expression with all patients (8/8) assigned AKR1C3high (Supplementary Fig. S15) [33].
We evaluated ex vivo ACHM-025 activity against a subset of PDX samples that were established from patients with acute myeloid leukemia (AML), neuroblastoma (NB), Ewing sarcoma (ES), diffuse midline glioma (DMG), hepatoblastoma (HB), and ependymoma (EPD) (Supplementary Fig. S16). Interestingly, AKR1C3 expression of PDX samples (as measured by qRT-PCR) did not reflect AKR1C3 expression of matched patient samples (as measured by RNA-seq) for NB or DMG (Supplementary Fig. S17A–C, Supplementary Table S13). For this reason, PDXs were assigned AKR1C3high based on qRT-PCR data of the PDX samples (CNRQ > 7; Fig. 6G, Supplementary Table
留言 (0)