
記住我

Core binding factor acute myeloid leukemia (CBF-AML), characterized by the recurrent translocations of t(8;21)(q22;q22) or inv(16)(p13.1q22)/t(16;16)(p13.1;q22), hereafter abbreviated as t(8;21) and inv(16), is associated with favorable outcome and response well to high-dose cytarabine-based therapy [1, 2]. Approximately 15% of CBF-AML cases are therapy-related, typically arising after topoisomerase II inhibitor exposure. While the outcomes of therapy-related acute promyelocytic leukemia are comparable to de novo cases, therapy-related CBF-AML (t-CBF-AML) has variably reported inferior outcomes compared to de novo CBF-AML (dn-CBF-AML) [3,4,5,6], often influenced by factors such as older age, antecedent malignancies, and cumulative treatment toxicity [7]. The ELN 2022 defined myelodysplasia-related cytogenetics (MDS-cyto) and myelodysplasia-related gene (MDS-gene) mutations are commonly found in t-AML and are associated with adverse prognosis [7, 8], but their significance in t-CBF-AML is unclear. Secondary cytogenetic abnormalities (SCA) and gene mutations are also frequent in CBF-AML, though their prognostic significance remains controversial [3, 4, 9,10,11].
We retrospectively reviewed patients aged ≥18 years with newly diagnosed CBF-AML at our center between January 2013 to December 2022 to evaluate the outcomes and genetic profiles between dn-CBF-AML and t-CBF-AML. The diagnosis of CBF-AML was confirmed by karyotyping, fluorescence in situ hybridization, or detection of RUNX1::RUNX1T1 and CBFB::MYH11 by reverse transcription-polymerase chain reaction (RT-PCR). Measurable residual disease (MRD) monitoring was performed using RT-qPCR at post-induction, post-consolidation, and every 3 months for 24 months. The definitions of MDS-cyto, MDS-gene mutations, molecular and hematological relapse are as per ELN 2022 (Supplementary Information) [12]. Note we defined complex karyotype (CK) as ≥3 unrelated chromosome abnormalities, including t(8;21) or inv(16) with at least one monosomy and an additional structural abnormality.
Event-free survival (EFS) events included molecular or hematological relapse or death. Cumulative incidence of hematological relapse (CIHR) was calculated using the gray test, with death in remission as a competing risk. Univariate Cox proportional hazard models estimated unadjusted hazard ratios (HR) for baseline factors, with variables showing P < 0.10 considered for multivariable analysis (MVA). Data analysis was conducted using EZR software [13].
This study was conducted in accordance with the Declaration of Helsinki and approved by the UHN Research Ethics Board (23-5164). All patients gave informed consent to be included in the institutional leukemia database, from which the study data were derived.
A total of 136 CBF-AML patients were identified, of which 25 (18.4%) had t-CBF-AML. Twenty-seven patients (19.9%) had prior history of cancers, with lymphoma (n = 8, 29.6%) and breast (n = 7, 25.9%) cancer being the most common. Therapy-related patients were older (median age of 64 vs. 48 years in dn-CBF-AML, p = 0.001) and predominantly female (68% vs. 36%, p = 0.03). Extramedullary disease was absent in t-CBF-AML but present in 13.5% of dn-CBF-AML cases. Hematologic parameters were similar between t-CBF-AML and dn-CBF-AML (Table 1).
Table 1 Comparison of clinical and genetic profiles between de novo and therapy-related cases of t(8;21) and inv(16) AML.SCA were seen in 50–60% of patients, with no significant difference in the cytogenetic profiles between de novo and therapy-related cases, including the rates of MDS-cyto. Only 2 patients with CK were identified, both in the dn-inv(16) group. Gene mutations were detected in 80–90% of patients across all subgroups. While MDS-gene mutations appeared more common in therapy-related cases, the differences were not statistically significant for t(8;21) (44.4% vs. 28.2%, p = 0.432) or inv(16) (33.3% vs. 11.6%, p = 0.130). We found no significant differences in the prevalence of chromatin modifier genes (BCOR, BCORL1, EZH2, KDM6A, ASXL2, ASXL1), cohesion genes (STAG2, SMC3, SMC1A, RAD21), DNA methylation genes (IDH2, IDH1, TET2), transcription factors genes (IKZF1, GATA2, RUNX1, ETV6, SETBP1, biallelic CEBPA), tyrosine kinase genes (PTPN11, JAK2, FLT3-ITD, FLT3-TKD, KRAS, NRAS, KIT, CBL) and tumor suppressor genes (WT1, PHF6) between de novo and therapy-related cases within each subset. Mutations frequently associated with therapy-related AML, including TP53, PTPN11 and PPM1D, were not found in our t-CBF-AML cohort.
Fewer t-t(8;21) patients received intensive therapy compared to dn-t(8;21) (60% vs. 91.1%, p = 0.024), or completed three or more consolidation cycles (25% vs. 84.3%, p = 0.001), likely due to older age (66.5 vs. 46 years, p = 0.111). Conversely, t-inv(16) and dn-inv(16) cohorts had similar rates of intensive therapy and consolidation cycles, despite the t-inv(16) cohort being significantly older (63 vs. 46 years, p = 0.001). Twelve patients (10.2%) received gemtuzumab ozogamicin (GO)—10 patients (9.8%) in the dn-CBF-AML group and 2 patients (12.5%) in the t-CBF-AML group. AlloSCTCR1 rates were similar between de novo and therapy-related cases (34% vs. 35.7%, p = 0.999). The primary indication for alloSCTCR1 in dn-CBF-AML was suboptimal molecular MRD clearance (44%). In t-CBF-AML, the primary reason was therapy-related history (57%), followed by suboptimal molecular MRD clearance (42.9%) (Supplementary Table 4).
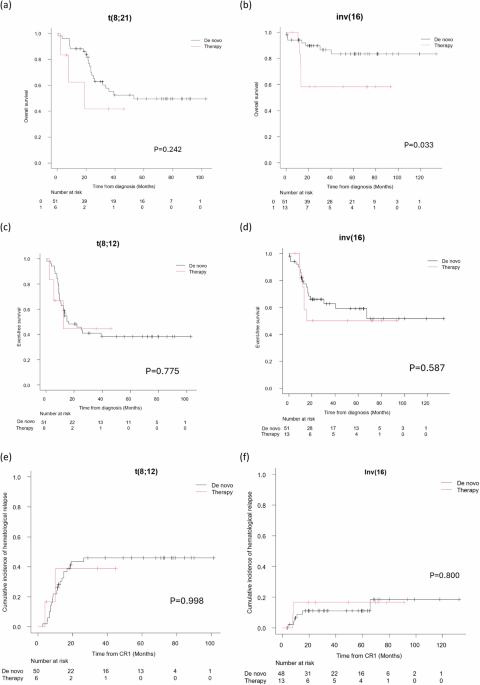
Post-induction (PI) complete remission (CR) was achieved in 97.5% of patients receiving intensive therapy. MRD data (Supplementary Table 5) was available for 65.2% at PI and 80.2% at end-of-treatment (EOT). The majority (80.4%) achieved MRD ≥ 3LR at EOT, with no significant difference in MRD response or relapse rates between de novo and therapy-related cases. The median follow-up time was 61.05 months (95% CI 48.72–66.23). Among intensively treated patients, the OS was worse in the t-t(8;21) cohort compared to the dn-t(8;21) cohort, with median OS of 18.96 months versus 53.13 months, respectively (p = 0.242). (Fig. 1a). The median OS for both dn-inv(16) and t-inv(16) was not reached, but 5-year OS was lower in t-inv(16) (58.3% vs. 83.2%, p = 0.033) (Fig. 1b). EFS did not differ significantly between de novo and therapy-related cases (Fig. 1c, d), and the two-year CIHR was similar between the de novo and therapy-related groups (Fig. 1e, f). Among patients receiving GO, two had hematologic relapse, and none experienced treatment-related mortality (TRM). Mortality was significantly higher in the t(8;21) cohort with disease progression being the primary cause of death (Supplementary Table 7). Conversely, TRM were the major cause of death in both de novo and therapy-related inv(16).
Fig. 1: Outcomes of de novo versus therapy-related t(8;21) and inv(16) cases.
Comparison of the overall survival (a, b), event-free survival (c, d), and cumulative incidence of hematological relapse (e, f) between de novo and therapy-related cases in t(8;21) and inv(16).
Among patients with EOT MRD ≥ 3LR, 68% achieved long-term remission without alloSCT, with 28.3% relapsed. Of these, 82.4% achieved CR2, with half undergoing SCT in CR2. AlloSCTCR1 was associated with a lower relapse rate (5%) but had a 10% TRM. Among patients with EOT MRD < 3LR, 54% relapsed without alloSCTCR1, and only 28.5% achieved CR2. AlloSCTCR1 reduced relapse (22%) with 11% TRM.
KIT mutations were associated with worse outcomes in t(8;21), particularly without alloSCTCR1. Among t(8;21) patients, the 5-year OS was 56.2% for KIT wild-type (KITWT) and 34.7% for KIT-mutant (KITMUT) (p = 0.0986), while the 2-year CIHR was 36.6% for KITWT and 54.4% for KITMUT (p = 0.174). After censoring for alloSCTCR1, the 5-year OS was 77.6% for KITWT vs. 16.3% for KITMUT (p = 0.006). MDS-cyto did not significantly impact OS, EFS, or CIHR, even after adjusting for alloSCTCR1. Both cases of CK achieved EOT MRD > 4LR and were alive and leukemia-free at the data cut-off. MDS-gene mutations were present in 23% of patients, who were generally older (median age 59 vs 46, p = 0.050) with a higher mutation burden (median number 3 versus 1, p < 0.001). None of the patients with MDS-gene mutations had antecedent myeloid neoplasms. There was a lower rate of intensive treatment in patients with MDS-gene mutation compared to those without (73.9% vs 93.4%, p = 0.018), although no significant difference in the rate of alloSCTCR1 (31.6% vs 26.1%, p = 0.772). In patients who received intensive therapy, the presence of MDS-gene mutations did not significantly affect survival or relapse rates.
On MVA, tyrosine kinase gene mutations were associated with worse survival (HR 3.2, p = 0.049) and higher relapse risk (HR 2.86, p = 0.018) in t(8;21). PI MRD < 3LR (HR 10, p = 0.027) was the strongest predictor for worse OS. For inv(16), older age (HR 1.11, p = 0.006) and EOT MRD < 3LR (HR 10, p = 0.010) predicted worse OS while alloSCTCR1 had a strong protective effect against relapse (HR 2.17e-5, p < 0.001).
While our study has limitations due to its single-center, retrospective design and small sample size, it supports findings from other studies indicating that host factors, such as age and treatment tolerance, primarily drive the worse outcomes in t-CBF-AML, rather than differences in disease biology [3, 5]. The genetic profiles of t-CBF-AML and dn-CBF-AML did not differ significantly, and the presence or absence of SCA did not significantly impact outcomes. MDS-cyto and MDS-gene mutations were not predictive of survival or relapse in our cohort. MRD status was a key predictor of outcomes, and alloSCTCR1 is recommended for patients failing to achieve MRD ≥ 3LR. Our MVA suggested that PI MRD had a stronger predictive value than EOT MRD in t(8;21), with the presence of tyrosine kinase gene mutations (majority were KIT mutation) serving as an additional high-risk feature. Importantly, there was no difference in MRD response between de-novo and therapy-related cases.
Despite the high CR1 rates, 40–50% of t(8;21) patients relapse, even after alloSCTCR1 (30% relapse post-transplant in our cohort). Patients with EOT MRD < 3LR who are ineligible for SCTCR1 also have high relapse rates and low CR2 achievement. Hypomethylating agents as maintenance therapy, supported by small trials, show potential promise in such cases [14, 15].
In conclusion, survival in t-CBF-AML patients is inferior to dn-CBF-AML, primarily due to host factors, rather than disease biology. The indication for alloSCTCR1 should be based on molecular MRD results rather than a history of therapy-related AML or MDS-associated mutations. PI MRD < 3LR and tyrosine kinase mutations predict poor outcomes in t(8;21), while EOT MRD < 3LR is the key predictive factor in inv(16).
留言 (0)