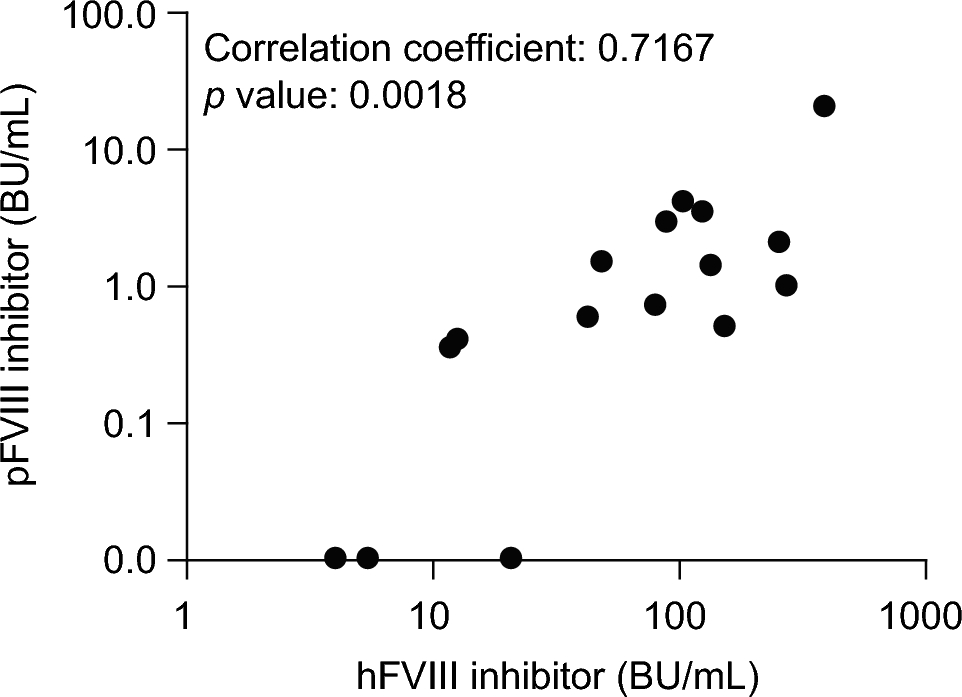
Since 2014, rpFVIII has been approved and used in clinical practice in the USA and Europe, and is positioned as a first-line hemostatic treatment for PwAHA alongside bypassing agents. One of the challenges of using rpFVIII products is the cross-reaction of anti-hFVIII autoantibodies in PwAHA. Analyses of the GTH-AH 01/2010 study showed that 44% of patient plasma samples cross-reacted to rpFVIII [12]. The results obtained in our study showed that rpFVIII elicited a cross-reaction in 56% of plasma samples from PwAHA, similar to the GTH-AH 01/2010 data. In addition, our study showed a median cross-reactivity of 1.2% (IQR 0.3–3.1%, range 0–5.1%), similar to the cross-reactivity reported in the GTH-AH 01/2010 study (0%; IQR 0–3.2%, range 0–46.7%). Together, these results indicate that the cross-reactivity of PwAHA autoantibodies to rpFVIII is similar in Japanese and Caucasian patients. In the present study, there was only one patient with a pFVIII inhibitor titer equal to or above 10 BU/mL, at 19.9 BU/mL. The inhibitors in each of the other patient samples could theoretically be neutralized with 200 U/kg of rpFVIII, resulting in increased plasma FVIII:C to at least 100%. This suggests that the initial clinical dose of rpFVIII could neutralize the cross-reactive pFVIII inhibitors found in the plasma of most PwAHA.
Data from the GTH-AH 01/2010 study suggest a tendency for greater cross-reactivity in samples containing autoantibodies against epitopes in the C1 domain [12]. In our study, cross-reactivity tended to be higher in the samples with epitopes only on the light chain (A3-C1-C2) than in the samples with epitopes only on the heavy chain (A1-A2), which is consistent with previous reports [26]. However, there were cases in which no epitope was detected, or specific epitopes were not identified. This made it difficult to verify the difference in cross-reactivity, owing to the difference in epitopes compared with the GTH-AH 01/2010 data.
FVIII:C levels exceeded 100% in all patient samples when plasma was spiked with 200 U/kg of rpFVIII (5 U/mL). Unexpectedly, FVIII:C was 412% in a sample with a pFVIII inhibitor titer of 19.9 BU/mL (patient 7). Initially, there was no correlation between FVIII:C and pFVIII inhibitor titer after addition of rpFVIII. However, upon exclusion of this sample (patient 7), a weak negative correlation without statistical significance was observed. Therefore, the pFVIII inhibitor value for patient 7 was considered an outlier. Compared with the present study, in a previous study in which the baseline pFVIII inhibitor titer ranged from less than 0.6 BU to 29 BU, there was a more pronounced negative correlation between pFVIII inhibitor titer and FVIII:C increase in patients after administration of rpFVIII [11]. However, in the present study, only one patient had a pFVIII inhibitor titer above 5 BU/mL, and this was the patient who was considered an outlier. Therefore, it is difficult to directly compare the results between the two studies. Autoantibodies in PwAHA are known to behave in a type 2 manner [19, 27], in that some endogenous FVIII:C remains even when FVIII inhibitor titer is high. Although cross-reactivity was low in our study, as indicated by the pFVIII inhibitor titer determined by the Bethesda assay, it is possible that the pFVIII inhibitor titer and FVIII:C after administration of rpFVIII behave as type 2, suggesting the influence of non-neutralizing antibodies (binding antibodies). Therefore, the recovery in FVIII:C after rpFVIII treatment is difficult to predict based on the pFVIII inhibitor titer. Rather, measuring of FVIII:C should be used for the monitoring of rpFVIII hemostasis as recommended in the previous report [11]. It should also be noted that FVIII:C was only measured at one time point (30 min) after rpFVIII addition in our study, owing to limited sample volumes. The time-dependent FVIII:C inhibitory effects of autoantibodies (e.g., at 1 or 2 h after rpFVIII addition) should be considered in future research.
Previous reports have demonstrated the utility of TGA and CWA parameters in assessing endogenous comprehensive coagulation potential in PwAHA [28]. There have been no reports evaluating TGA and CWA in PwAHA after administration of rpFVIII; therefore, our study investigated whether these assays might be useful as a monitoring index in this context. All parameters from CWA and TGA were found to restore to levels similar to normal plasma after spiking with rpFVIII. In some patient samples, the parameter levels were low. In addition, there was no correlation of these CWA and TGA parameters with pFVIII inhibitor titers overall. Previous reports have shown that peak thrombin and time to peak had a strong inverse correlation with pFVIII inhibitor titers after rpFVIII addition to the plasma from PwCHA-INH [17]. In PwCHA-INH, inhibitor behavior is known to be predominantly type 1 [19, 27], and the inhibitor titer and FVIII:C show a linear negative correlation. Because the behavior of inhibitors in PwAHA is predominantly type 2 [19, 27] and, therefore, inhibitor titers may not correlate with FVIII:C values, it is reasonable that the comprehensive coagulation potential and the pFVIII inhibitor titer did not correlate in our study. However, the overall trend of recovery was similar to that of normal plasma, suggesting that the TGA results also support the clinical efficacy of rpFVIII.
The TGA and CWA parameters tended to be restored with increasing FVIII:C, but did not tend to increase markedly even with FVIII:C of over 200%. In our study, a dose equivalent to 200 U/kg of rpFVIII (final concentration: 5 U/mL) was added, and FVIII:C exceeded 200% in many samples. However, TGA and CWA parameters associated with increased FVIII:C reached a plateau, suggesting that the levels of other coagulation factors (e.g., FX, FIX, fibrinogen) or anticoagulants (e.g., protein S, protein C, anti-thrombin) in the plasma could be rate-limiting for TGA and CWA parameters. An increase in comprehensive coagulation potential beyond normal plasma was, however, observed in some samples. In the sample from patient 1, the CWA values were high for |min1| and |min2|, but not for Ad|min1| and Ad|min2| (see Supplementary Table S1), indicating the possibility that fibrinogen values may be related. The peak thrombin in TGA was also high in this sample. Considering that peak thrombin increases with increasing fibrinogen level [29], the possibility that patient fibrinogen levels were related to the high thrombin peak cannot be ruled out. In the sample from patient 5, FVIII:C increased up to 179% but TGA peak thrombin was low, while CWA |min1| and |min2| values were equivalent to control samples (see Supplementary Tables S1 and S2). In this sample, extrinsic factors (e.g., high tissue factor pathway inhibitor) rather than intrinsic factors seemed likely to be involved. Although TGA parameters recovered to levels comparable to normal plasma overall, caution may be needed because it was observed that some patients had insufficient recovery of peak thrombin when compared with the mean [SD] peak thrombin observed in control plasma (234.8 [17.7] nM). The sample from patient 11 had very low |min1| and |min2|, but only a slightly low value for peak thrombin was observed, and Ad|min1| and Ad|min2| were similar to those of the control (see Supplementary Tables S1 and S2). Therefore, the influence of low fibrinogen levels should be considered. Thus, the possibility that some patients receiving a dose of 200 U/kg of rpFVIII may have different hemostatic effects in clinical practice cannot be denied. However, the patient characteristics and factors related to this differential response failed to be identified in our study. A study involving a larger number of patients in a clinical setting would be needed to investigate this.
Overall, despite some variability in responses to rpFVIII between individual patient samples, the results of coagulation potential for most samples in this study were within the expected range, and the results largely support the efficacy and safety of rpFVIII. Given that TGA and CWA parameters could depend on the activity of coagulation factors other than FVIII, or on anticoagulants in the patient’s plasma, further investigation is still needed to determine whether TGA and CWA are appropriate monitoring tests for evaluating the hemostatic potential of rpFVIII. In our study, there were no samples in which the FVIII:C was less than 100% after spiking with 200 U/kg of rpFVIII, although this could occur in clinical use. Monitoring of FVIII:C can detect a failure to increase the coagulation potential as expected, and appropriate actions such as additional administration of rpFVIII should be taken. For the proper use of rpFVIII, monitoring of FVIII:C and clinical symptoms, and appropriate adjustment of regimens are important.
A limitation of this study was that it was not an in vivo study, but rather an in vitro study using plasma from PwAHA. Apart from FVIII, no other coagulation factor levels, such as FX, FIX or fibrinogen, could be determined owing to limited sample volumes. In addition, there were a small number of samples, and only 200 U/kg of rpFVIII was used for investigation of coagulation potential. Owing to the small sample size, no tests could be performed to statistically compare rpFVIII cross-reactivity between Japanese and Caucasian patients for equivalence. Nevertheless, our findings demonstrate that the cross-reactivity to rpFVIII in Japanese PwAHA is similar to that reported in Caucasian patients. In addition, our results suggest that an initial clinical dose of rpFVIII 200 U/kg can restore coagulation potentials to normal levels. Monitoring of FVIII:C after administration of rpFVIII may be more informative than measuring pFVIII inhibitor titer before administration of rpFVIII.


留言 (0)