The idea that patients with immunodeficiency to B-CLPD may suffer from a late-onset PID is an increasingly on-date debate. Due to clinical similarities of both entities, a mild-symptomatology spectrum and the absence of a thorough clinical immunodeficiency-focused examination, many PID could be hidden until debuting with a malignant lymphoproliferative disorder. Indeed, PID are associated with higher risk of developing hematological malignant processes than general population [2, 20]. Therefore, identifying late-onset PID among patients with B-CLPD and immunodeficiency could have major importance for early infection prevention, and ultimately, to aim for a better prognosis with more targeted or adjusted therapies, beyond current protocols, when available, to potentially improve prognosis and reduce iatrogenic effects. For example, certain mutations in the context of APDS or Familial Hemophagocytic Lymphohistiocytosis (FHL) could be treated with specific targeted therapies or protocols, but most genetic variants do not currently have targeted therapies. In this work, we found 10 clinical and immunological variables that discriminated between “Suspected-PID” and SID patients. Furthermore, through a robust tree decision model, two of those variables: “sum κ + λ” and “childhood infections”, identified PID with high accuracy.
In our cohort, according to the model, 70% of the patients that were thought to be SID could be reclassified as “Suspected-PID”. We acknowledge that the initial 70% prevalence of ‘Suspected PID’ might appear disproportionate given the known prevalence rates of PID versus SID. However, the initial classification was based on rigorous clinical and immunological criteria of CVID diagnosis as outlined by the ESID Registry [13]. This classification may overlap SID patients with infectious manifestations years prior to the overt cancer. The relevance of our AI regression algorithm is that it did not rely solely on these criteria. Instead, it selected a combination of vertical criteria (childhood recurrent or severe infections) and horizontal criteria (undetectable sFLC) to give a more consistent approach that may indicate a cohort skewed towards hidden PID, underscoring the complexity of differentiating between PID and SID and stressing the critical need for a comprehensive clinical and immunological evaluation at the time of B-CLPD diagnosis. The observed high prevalence of suspected PID might be influenced by a referral bias, as hematologists often refer patients who present with significant infectious complications.
Even though the number of genetic variants identified to be related to PID pathogenesis is increasing exponentially, to date, PID diagnosis still relies heavily on clinical criteria. Likewise, in our study, the weight of recurrent or severe infections early in life was the second best classifier. We also acknowledge the potential importance of certain clinical variables, like the increased frequency of autoimmune disorders and malabsorption [20, 21] in the “Suspected PID Group”. While these findings did not reach statistical significance, this may be attributed to our limited sample size. These conditions are traditionally linked to PID, which can lead to poorer outcomes and an increased risk of neoplasia [5]. Nonetheless, they should be further tested in larger cohorts to verify their potential diagnostic role.
Our study revealed that patients in the “Suspected-PID Group” not only exhibited higher rates of complete remission but also demonstrated greater requirements of Rituximab for cancer control, and of IgRT for infection prevention. Altogether, this suggests that these patients may exhibit a different clinical behavior due to higher immunogenicity while also being more vulnerable to immunodepletion and infection-related complications. In response, their treatment protocols may necessitate adjustments or the incorporation of targeted therapies. Conventional aggressive anti-tumor therapies could exacerbate the immune state of “Suspected-PID” patients, heightening their susceptibility to infections and subsequent organ damage, aligning with the increased demand for IgRT observed in our research. Historically, cancer treatments primarily aimed at reducing malignant cell populations. Recently, attention has shifted towards reverting the immunosuppressed tumor microenvironment with novel immunotherapies. Nonetheless, the potential systemic immunodeficiency in patients has not been sufficiently considered. Understanding the variables that distinguish these groups can help tailor diagnostic and treatment strategies to the specific needs of patients, potentially leading to better outcomes.
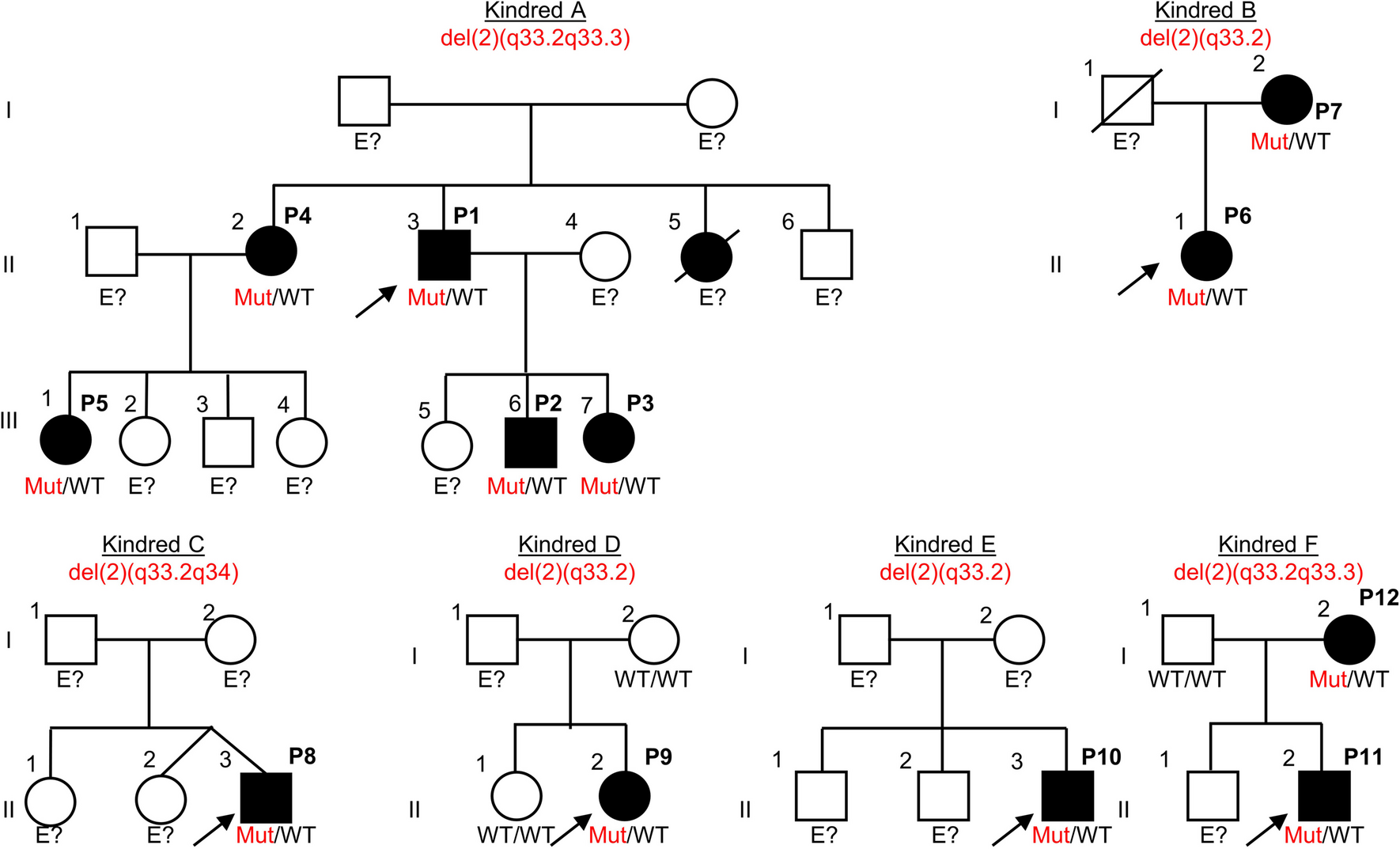
An intriguing and original finding was the presence of a family history of lymphoid malignancies, which is challenging to interpret, and raises the possibility of an enrichment of other genetic defects associated with cancer, rather than directly supporting a PID diagnosis. However, this observation could also point to the presence of driver germline variants within the family, particularly at the intersection between PID and cancer/lymphoproliferation. A significant proportion of genetic variants related to IEI, including those in our cohort, are linked to a higher susceptibility to lymphoproliferation. Based on this, we hypothesize that a higher frequency of familial lymphoid malignancies may indicate an underlying genetic defect contributing to PID and cancer, though this remains speculative and requires further investigation in larger cohorts and additional studies.
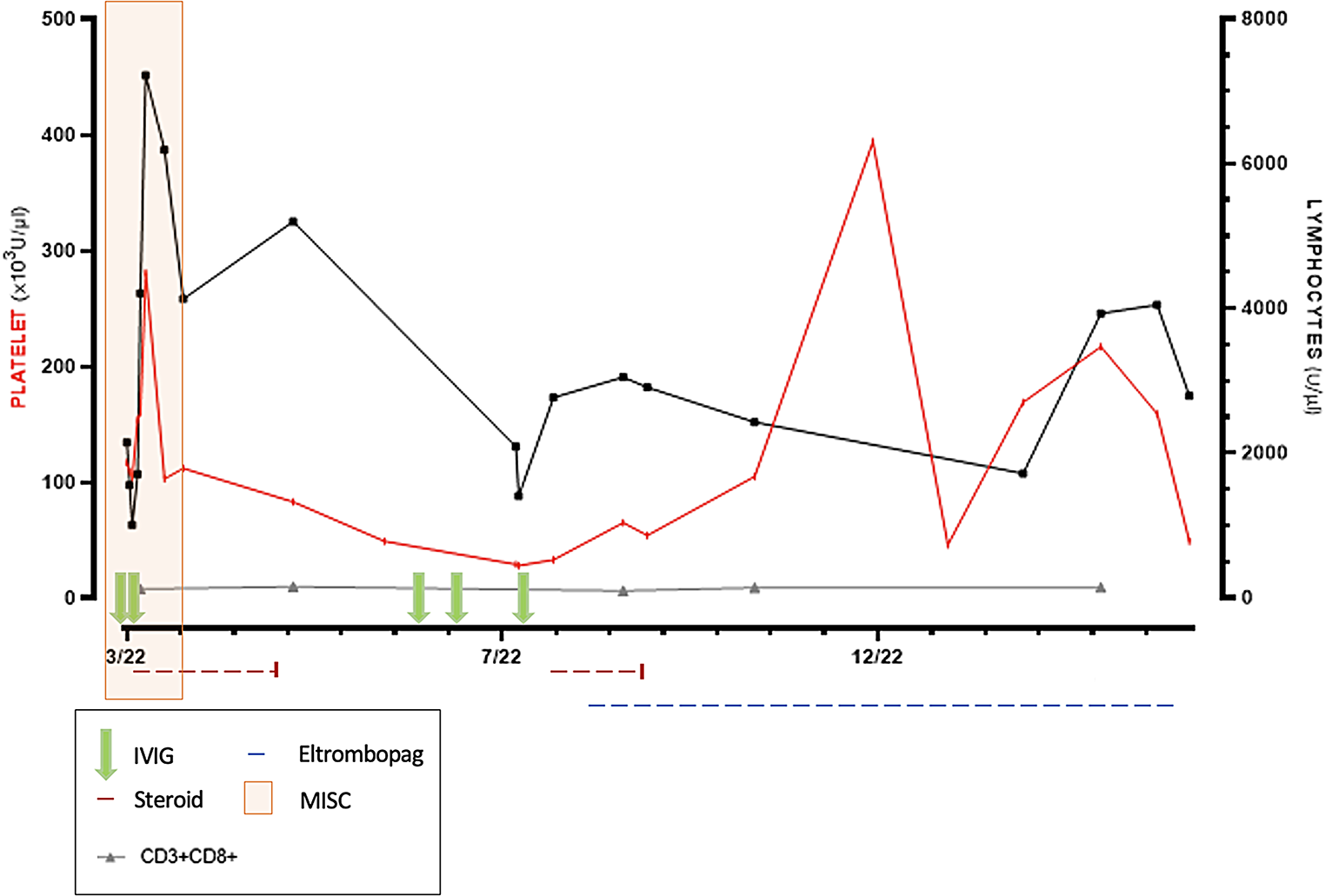
When considering analytical biomarkers, very few of them have been proved to discriminate between PID diagnoses yet. sFLC, and specifically, sum κ + λ, however, may serve as a valuable biomarker in distinguishing between primary and secondary hypogammaglobulinemia, particularly in the context of CVID versus SID [17, 22, 23]. In our study, we found statistically significant differences in sFLC κ and λ concentrations, and in sum κ + λ between “Group Suspected-PID” and SID patients, and in NHL subgroup analysis, which could validate the use of sum κ + λ in SID populations. Serum concentration of λ was more significant than κ concentration, probably due to clonality tends to be more frequent in κ chain. During normal B-cell development, κ is produced in higher quantities than λ. Although not yet demonstrated, it could also be possible that, in impaired B-cell development, λ production is affected earlier, making it a more sensitive biomarker than κ. Interestingly, the sum κ + λ measurement was taken after patients had undergone anti-tumor therapies, including anti-CD20 therapies, with extended follow-up, particularly in the subanalysis of NHL. Despite this, the “Suspected PID NHL” maintained very low concentrations.
One potential limitation that reflects the real-world experience is that 61.76% of “Suspected-PID Group” vs. 38.24% of SID had been on an anti-CD20 therapy, making it difficult to discriminate the cause of the underlying B-cell defect. Normal B-cell reconstitution is described to occur from 6 to 9 months to, approximately, 2 years after therapy [24]. Most suspected PID patients failed to achieve B-cell reconstitution even after a decade, suggesting a potential intrinsic B-cell defect potentially triggered by prior therapies, as hypothesized for other conditions [25,26,27]. This persistent deficit may have disrupted the normal B-cell reconstitution process, resulting in sustained small B-cell depletion and below range serum κ + λ levels. However, even though these results should be verified in further studies, we believe that testing B-cell parameters in these patients before undergoing therapy could be essential for their identification.
Despite these patients share some similarities, just as it is known about PIDs, the great variability of their clinical and immunological presentations can make it a very challenging task to reach a suited diagnosis, and even more, when they present with the diagnosis of a malignant disorder. In that sense, AI tools could aid clinicians reduce the “noise” of the vast clinical spectrum to reach an early diagnosis. We developed an AI model of diagnosis which is based on the most significant variables creating a regression tree decision model that seems to accurately target patients with a high suspicion of late-onset PID at time of malignancy diagnosis. Three quarters of our cohort could be identified as possibly having a PID with this model, highlighting the importance of identifying late-onset PID in the setting of B-CLPD with the help of AI. Unlike conventional statistical methods, the AI approach does not rely on assumptions about data distribution, allowing for a more robust analysis. AI went deeper into variable interactions, handled unobserved constructs by selecting a regression tree model out of different models. It also addressed measurement errors and conducted confirmatory factor analysis using bootstrapping for internal validation.
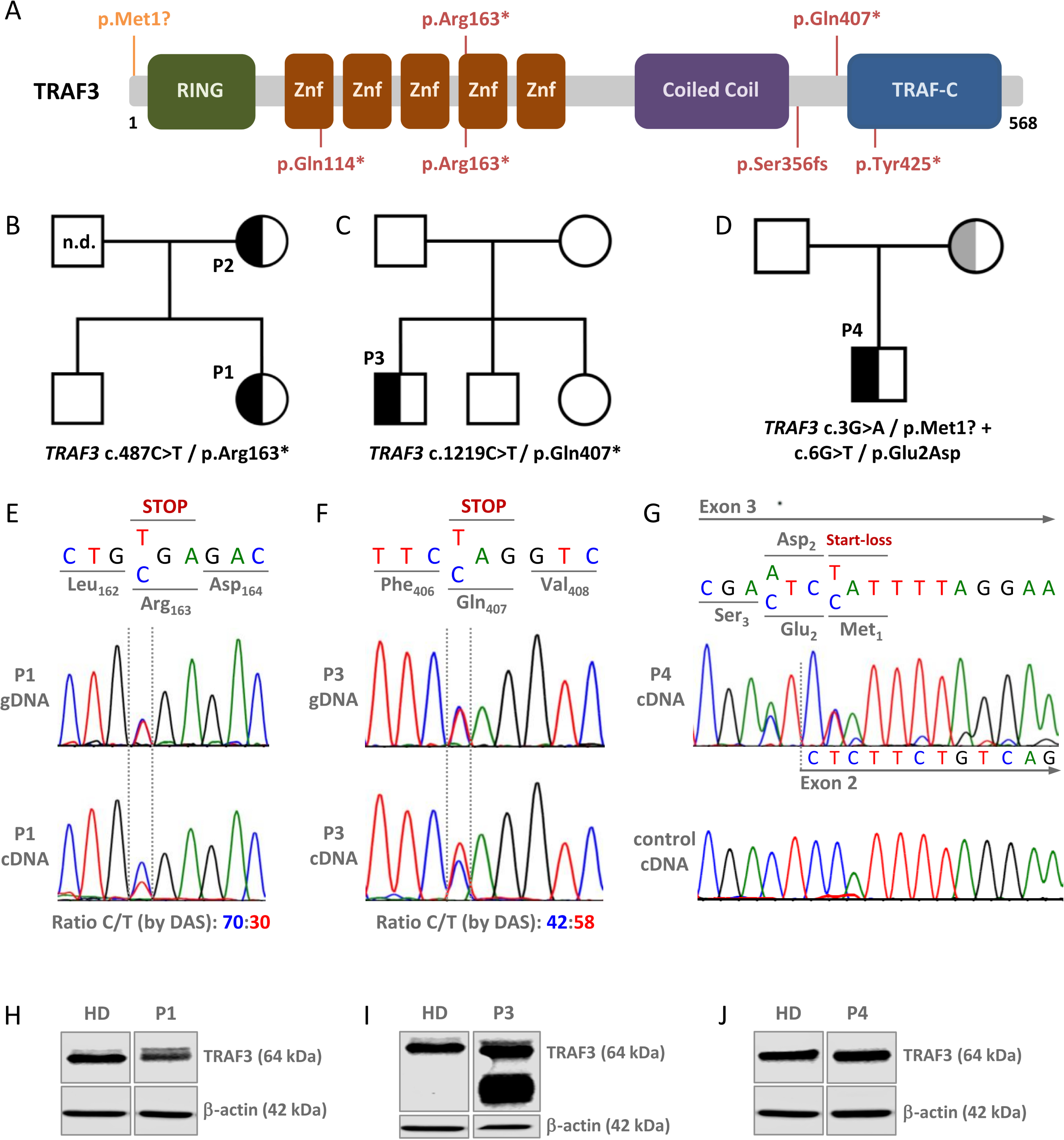
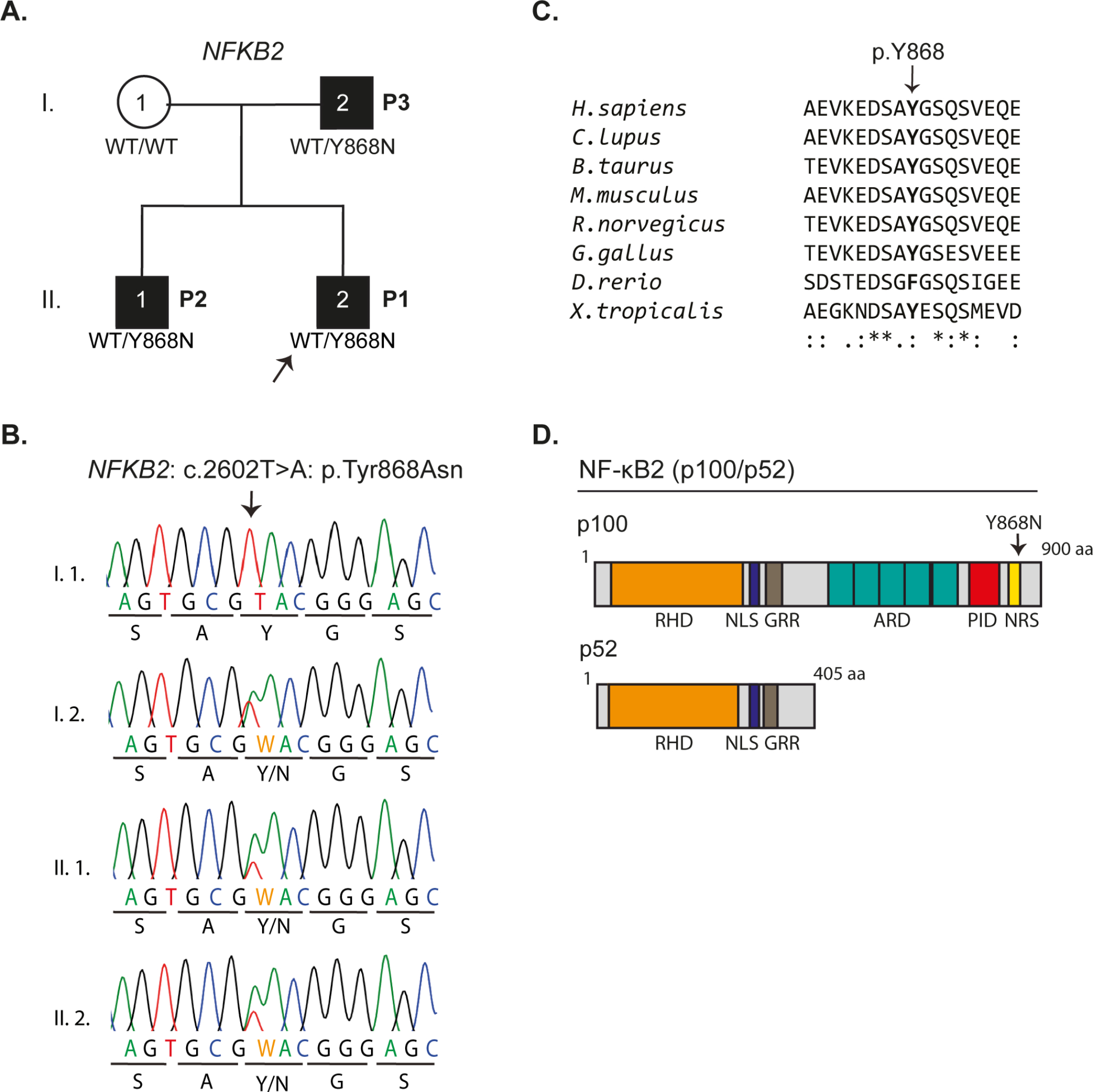
Accordingly, AI could help address genetic studies in those PID-suspected patients, allowing the study of genetic variants susceptible of more targeted therapies. In this study, we found that a high proportion up to 66.10% of 59 “Suspected-PID Group” patients carried, at least, one genetic variant related to IEI, according to Tangye SG et al. [1], supporting the hypothesis that PID and SID may fall below the same genetic and clinical spectrum. More than a third of them were related to combined immunodeficiencies, followed by immune dysregulation, and antibody deficiencies. Moreover, almost 20% of the variants related to combined immunodeficiencies and predominant antibody deficiencies, were also implicated in defects of DNA repair mechanisms. In line with that described for lymphomagenesis mechanisms in PID [4, 28], our data point to shared genetic defects related predominantly to immunosurveillance, intrinsic DNA repair and to B-cell biology intersecting PID and B-cell lymphoproliferative disorders. A broad spectrum of variants was observed, involving major signaling pathways that can impede the normal clearance of microorganisms and transformed cells, disrupt antibody production, and contribute to increased genetic instability and mutational load, as well as to chronic inflammation and lymphoproliferation. This underscores the intertwined nature of immunodeficiency and immune dysregulation and B-cell cancer. However, according to current literature, the heterozygous nature of the variants alone might not fully explain the clinical outcomes we see in our patients. Nonetheless, it is plausible that the delayed clinical presentation of the malignancy may be attributable to the heterozygosity of the genetic variants, possibly suggesting that these germinal genetic predispositions may require the cumulative effects by added somatic mutations and environmental triggers in a multi-hit process to manifest clinically [29]. It is likely, that if these variants were homozygous, patients might have been diagnosed earlier in life with classical pediatric IEI. CVID, the most common adult PID, can manifest from adolescence to late adulthood. While the prevalence of monogenic causes in CVID patients is estimated to be 25–30% [30], most cases could involve digenic or oligogenic variants, genetic modifiers, epigenetic factors, or somatic mutations acquired over time. Interestingly, the array of monogenic variants linked to CVID-like disorders is broad, encompassing over fifty variants that produce a similar clinical and immunological phenotype [31]. These variants also demonstrate significant overlaps with the somatic variants of NHL [32]. We argue that similar mechanisms might apply to our “Suspected-PID” patients, which could account for the presence of only heterozygous variants. Indeed, we could reinforce this idea by comparing those findings to the genetic results of our definite PID patients. Just as the “Suspected-PID” group, we found that a high percentage of the PID patients (64.71%) carried, at least, one genetic variant, being 94.44% of them present in genes related to IEIs. All of them were heterozygous as well, and also, similarly, they mostly belonged to the categories of ‘Predominantly antibody deficiencies’ and ‘Combined Immunodeficiencies’, with or without syndromic features. On the contrary, ‘Autoinflammatory disorders’ were more represented, and variants related to ‘Immune dysregulation’ were less observed than in the “Suspected-PID”. Interestingly, variants of ‘Combined Immunodeficiencies’ were more frequent in the “Suspected-PID” than in definite PID, possibly suggesting a higher risk of malignancy development due to defects in cellular immunosurveillance, further suggesting our hypothesis regarding family history of cancer malignancy.
It should be stated that a significant limitation of our study is the availability of genetic data exclusively from the “Suspected-PID” group with none from the “SID” group as comparison. However, these results stress that the identification of pathogenic variants confirming PID in previously SID patients could be of paramount importance in applying more personalized management [6]. It is imperative to accurately identify these patients at diagnosis to tailor personalized therapeutic strategies and follow-up plans, ensuring a holistic approach that addresses both the oncological and immunological challenges. Some examples of a more personalized management could be: the inclusion of periodic viral load tracing, as viral infections may worsen their prognosis and increase the risk of more malignant transformations in certain PID patients; specific therapy protocols, as for FHL or even specific targeted therapy in cases of APDS. Also, familial counseling should be included in those cases where genetic variants are identified. An intriguing observation from our study is the heightened familial incidence of hematological malignancies, particularly in the NHL subgroup among suspected-PID patients. This suggests that earlier detection of genetic variants could expedite the diagnosis of patients and potentially reduce the incidence of B-CLPD in cases involving IEIs and late-onset PID.
Beyond the immediate clinical scope, the distinctions among the groups based on specific variables point to opportunities for further research into the mechanisms driving these differences, as well as the development of new diagnostic tools or therapeutic approaches.
Overall, the decision tree model’s insights into the variables that discriminate among “Group PID”, and “Group SID” highlight the complexity of the cohort’s health profiles and the need for detailed analysis at time of lymphoproliferative diagnosis to support effective patient diagnosis, treatment and, hopefully, aid in their prognosis.



留言 (0)