
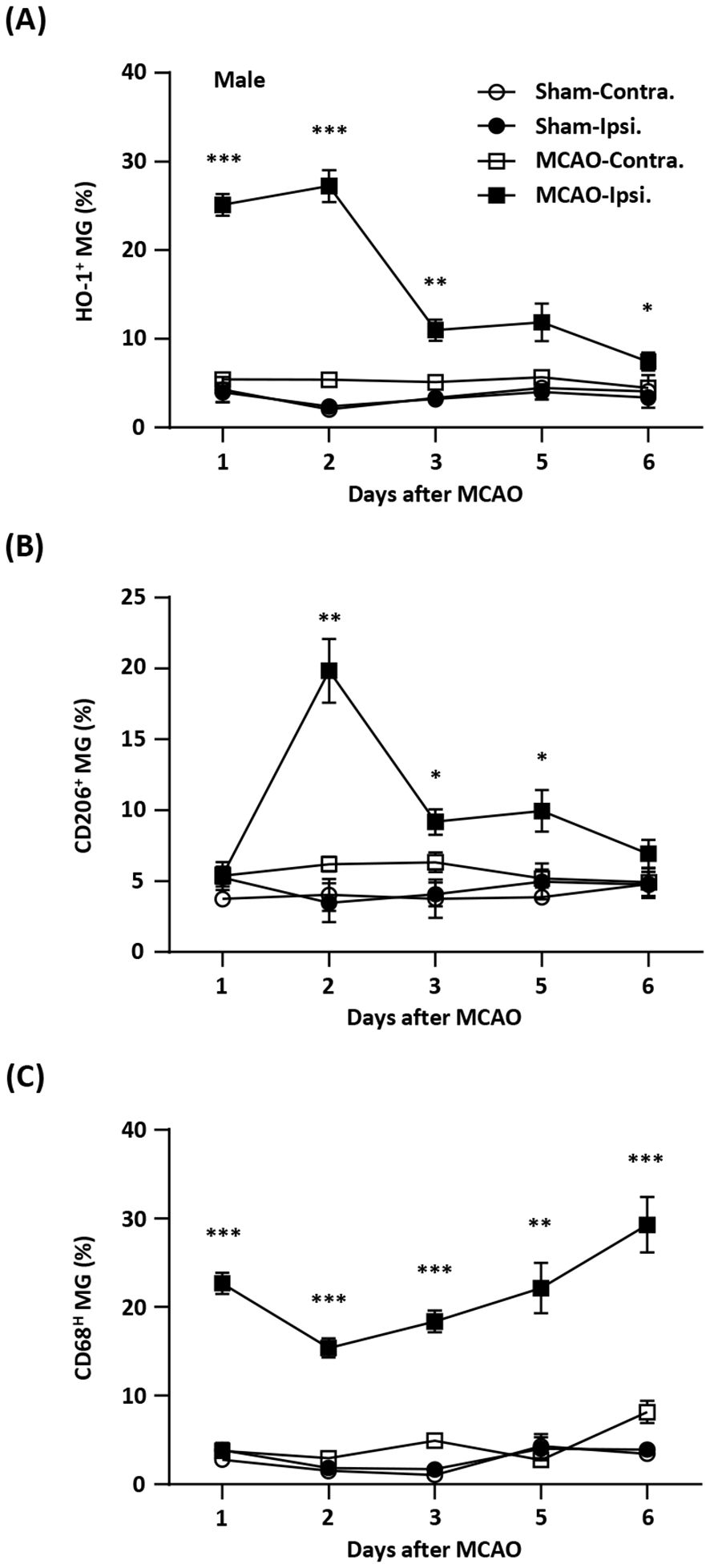
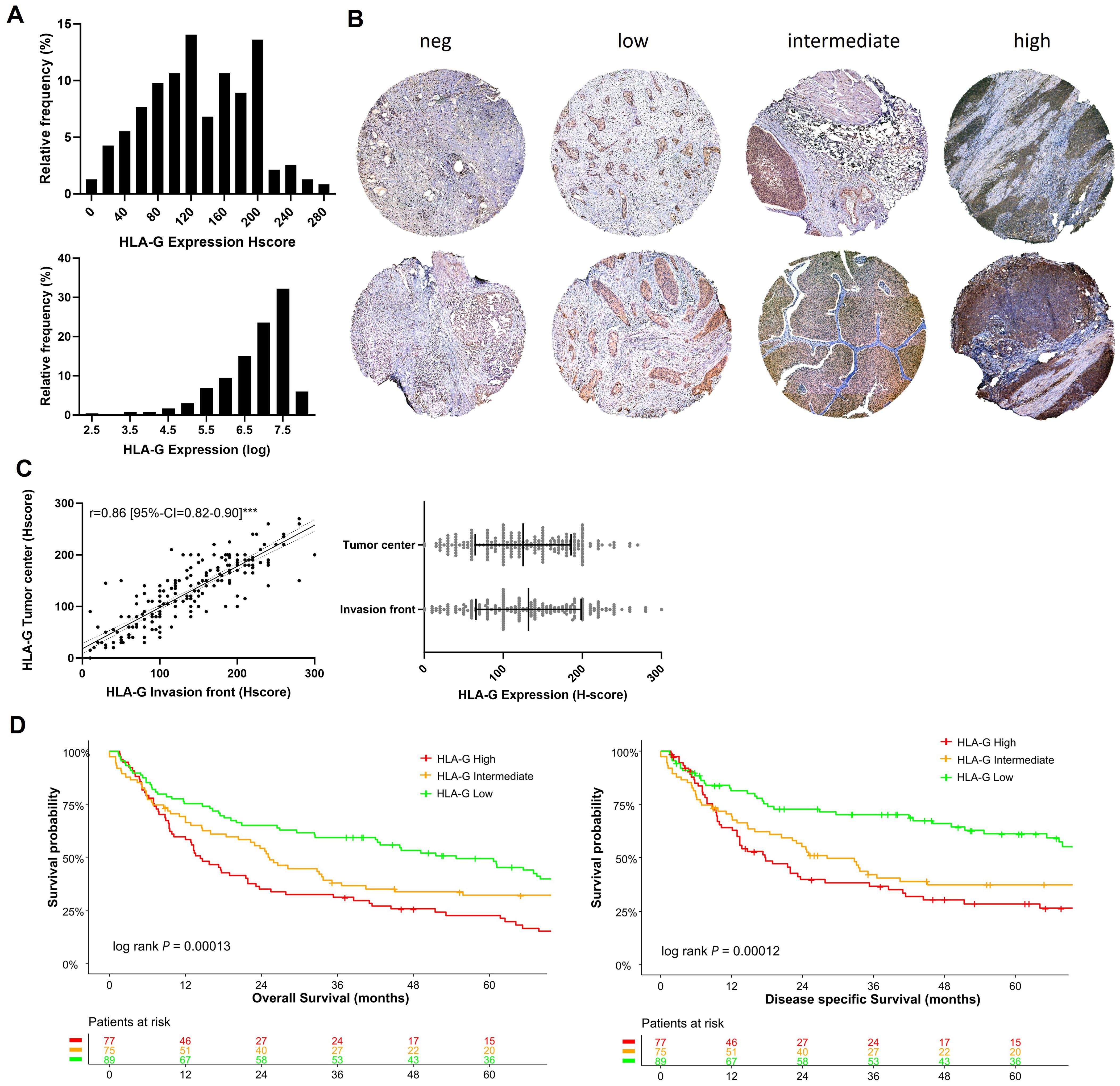
記住我
In vertebrates, red blood cells (RBCs) are abundant in the circulation and are the main medium for oxygen transportation in the blood. In recent years, several studies have demonstrated that erythroid cells have additional functions beyond oxygen transport. Given their high level of production, vast numbers, and whole-body distribution, understanding of the immunomodulatory effects of erythroid cells has potential to provide novel targets for future immunotherapy approaches.
The immunoregulatory effects of erythroid cells were first discovered over 70 years ago, in 1953, when Nelson RA Jr discovered the phenomenon of immune-adherence between microorganisms and erythrocytes, which caused an immunologically specific reaction and enhanced phagocytosis (1). Subsequently, in 1979, the immunosuppression mediated by splenic nucleated erythrocytes was first revealed (2), followed by the work of Conway de Macario in 1980, linking immunosuppression with erythropoiesis in irradiated spleen-cell-transferred C57BL/6J mice (3). These studies revealed that nucleated erythrocytes can suppress primary and secondary antibody-mediated responses in vivo (4). A few years later, nucleated erythrocytes, which inhibit B-cell proliferation in humoral immune responses, were named erythroid immunosuppressor cells (5). Recent studies have demonstrated that erythroid cells modulate both innate and adaptive immune responses (6). The aims of this review were to introduce the basic features of erythropoiesis and to summarize the immunomodulatory functions of RBCs and CD71+ erythroid cells (CECs).
2 ErythropoiesisErythropoiesis is a constant, multi-stage process, which takes approximately 14 days in adult humans, who produce almost 200 billion RBCs every day, while mice generate more than 7000 erythrocytes per second (7, 8). During adulthood, steady state RBC generation occurs in the bone marrow, while damaged and/or senescent RBCs are recognized, internalized, and digested by splenic red pulp macrophages and Kupffer cells in the liver. This cycle of production and clearance creates steady-state RBC life spans of approximately 120 and 60 days in humans and mice, respectively (9).
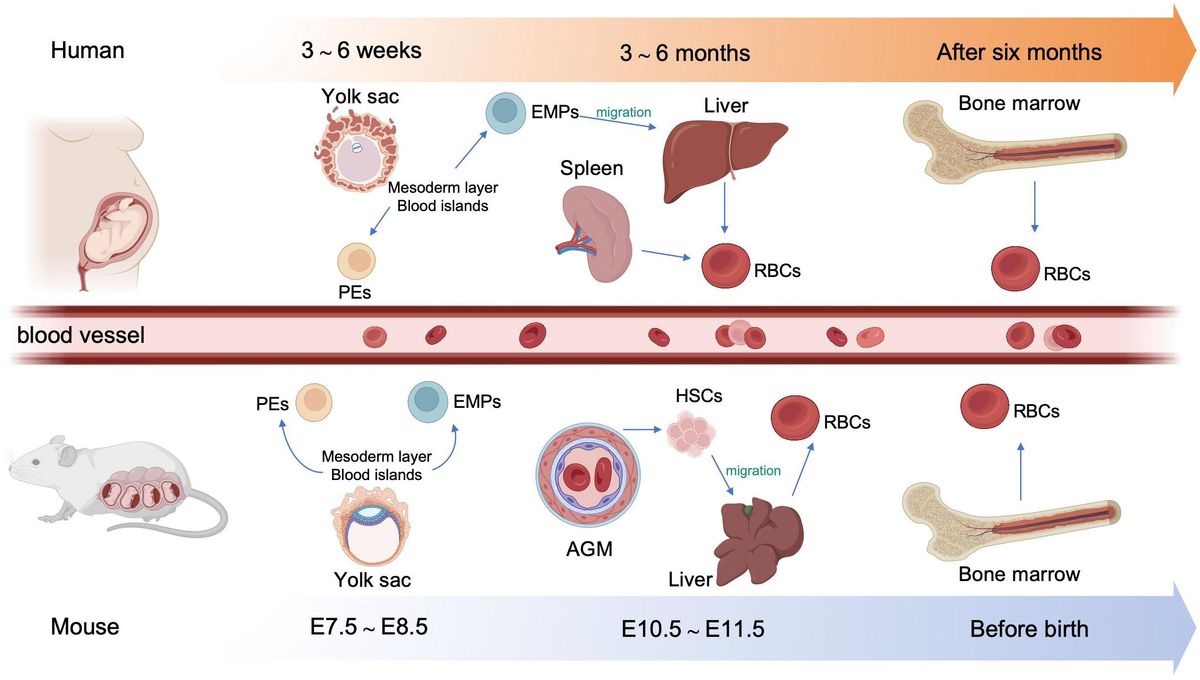
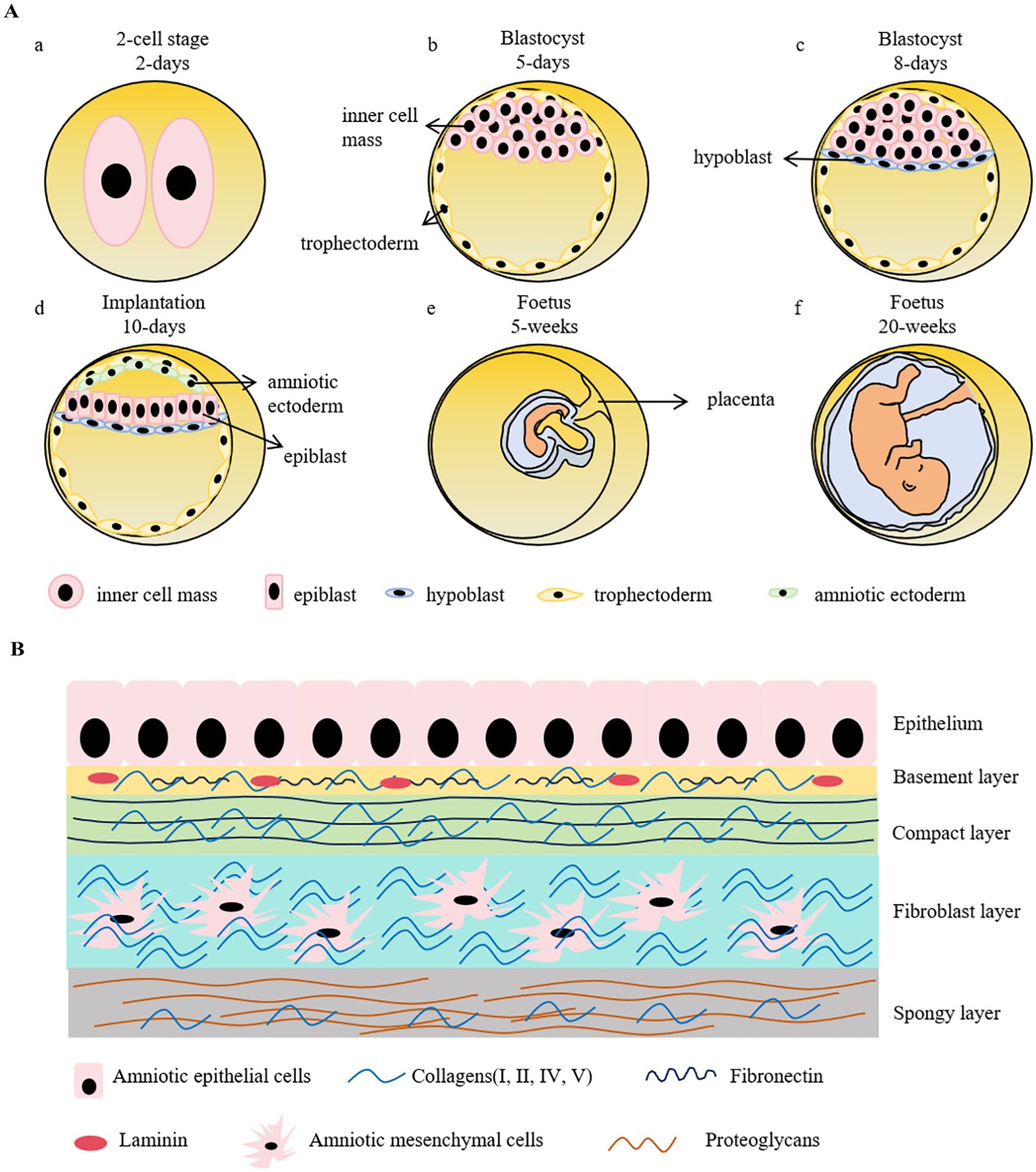
2.1 Developmental stages of erythropoiesis2.1.1 Embryonic hematopoiesisIn human embryos, erythropoiesis first occurs in the yolk sac, then transfers to the fetal liver and spleen, and finally becomes established in the bone marrow (10). Blood islands form from the mesoderm layer in the yolk sac, where primitive erythroid progenitor cells differentiate into primitive erythroblasts (PEs), which produce embryonic hemoglobin (α2ϵ2) (11). During weeks 6–8 of gestation, erythro-myeloid progenitors (EMPs) from the yolk sac begin to transfer to the fetal liver and spleen. The liver becomes the primary site of erythropoiesis during weeks 10–28 of gestation, while the spleen is the primary producer of RBCs during the second trimester (12, 13). At the end of the second trimester, erythropoiesis transfers to the bone marrow, which becomes the primary site of erythropoiesis until birth; fetal hemoglobin is produced to facilitate oxygen transport across the placenta during this stage (14). After birth, fetal hemoglobin output gradually decreases and is replaced by the adult form of hemoglobin. (Figure 1).
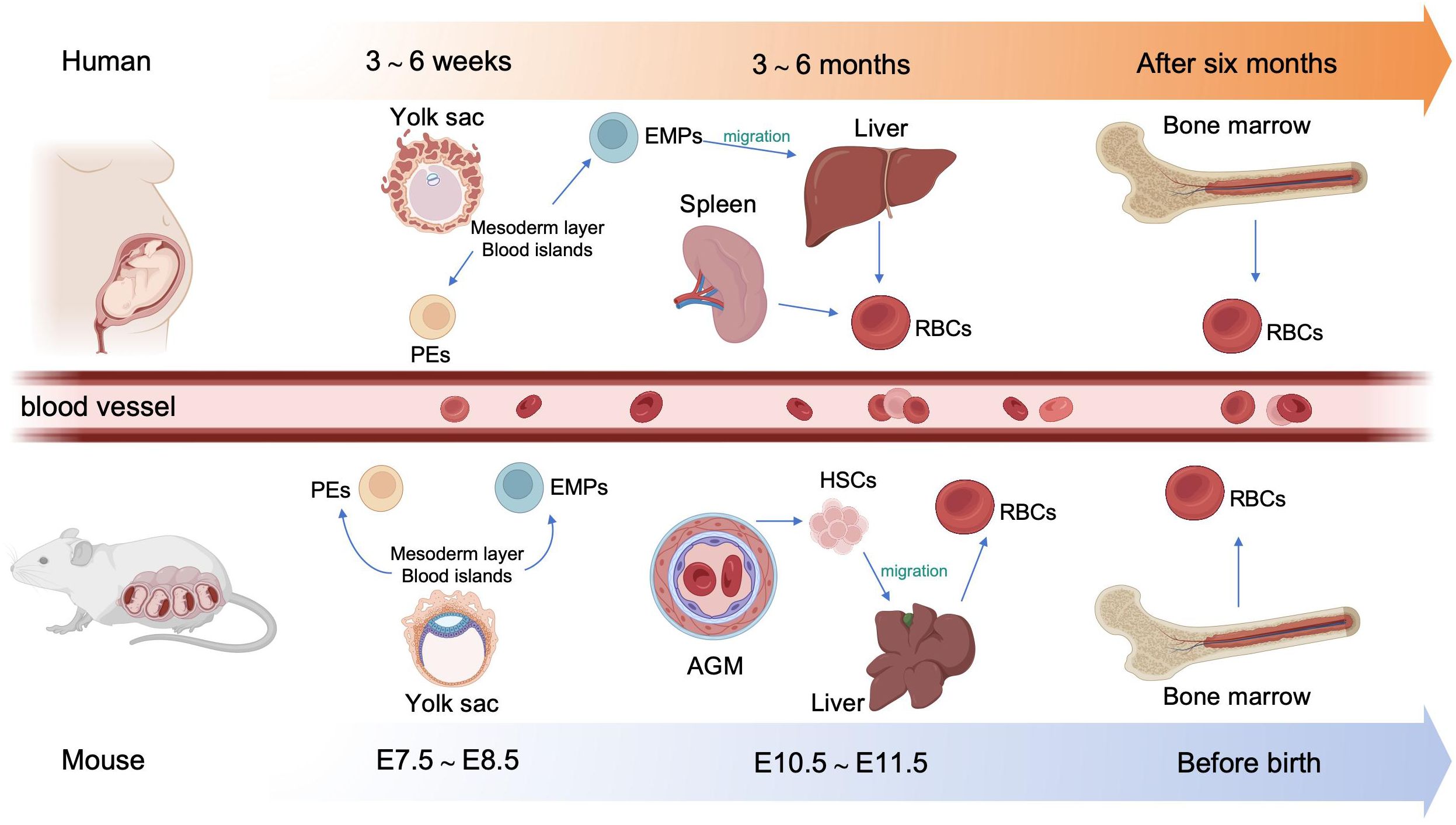
Figure 1. Overview of embryonic hematopoiesis in human and mouse. In human, erythropoiesis first occurs in the blood islands from the mesoderm layer of the yolk sac, generating primitive erythroblasts (PEs). Subsequently, erythromyeloid progenitors (EMPs) from the yolk sac migrate to the fetal liver and spleen. Finally erythropoiesis occurs in bone marrow. In mouse, primary erythropoiesis develops in the yolk sac. The yolk sac then atrophies and hematopoietic stem cells (HSCs) appear in aorta-gonad-mesonephros (AGM) region and transfer to the liver. Finally, erythropoiesis transfers to the bone marrow before birth.
In mouse embryos, hematopoiesis first emerges in the yolk sac at embryonic day 7.5 (E 7.5), and is characterized by the production of PEs, with diploid platelet progenitor cells and macrophages (15). Subsequently, EMPs emerge in the yolk sac at approximately E 8.25, which can generate erythroid colonies similar to those derived from adult bone marrow and have the capacity to produce multiple other myeloid lineages (16, 17). Soon afterwards, at around E10.5, hematopoietic stem cells (HSCs) appear in the dorsal aorta of the aorta-gonad-mesonephros region. Meanwhile, HSCs may also emerge from other hemogenic endothelial cells (ECs) within arteries in the umbilical cord, yolk sac, vitelline, cranial, and placental regions. These HSCs then migrate to the fetal liver, where they undergo a period of expansion, until they transfer to the bone marrow before birth (18) (Figure 1).
2.1.2 Stages of erythroid developmentThe development of erythroid cells during erythropoiesis can be divided into five stages. During the first stage, HSCs differentiate into megakaryocyte-erythroid progenitors (MEPs). The second stage is initiated by the differentiation of erythroid progenitor cells, followed by the appearance of burst-forming unit-erythroid (BFU-E) progenitors, and ends with the differentiation of colony-forming unit-erythroid (CFU-E) progenitors (19). The third stage begins with the development of pro-erythroblasts, followed sequentially by basophilic erythroblasts (Baso-E), polychromatic erythroblasts (Poly-E), and orthochromatic erythrocytes (Ortho-E). The fourth stage comprises reticulocytes, with mature erythroid cells formed in the fifth and final stage. Reticulocytes mature in the bone marrow, where they begin to eliminate mitochondria and other organelles, and subsequently enter the circulation to undergo further maturation into erythrocytes (20, 21). Erythroid cells gradually reduce their overall and nucleus size, while simultaneously increasing their hemoglobin content (10). Markers of erythropoiesis are listed in Table 1.
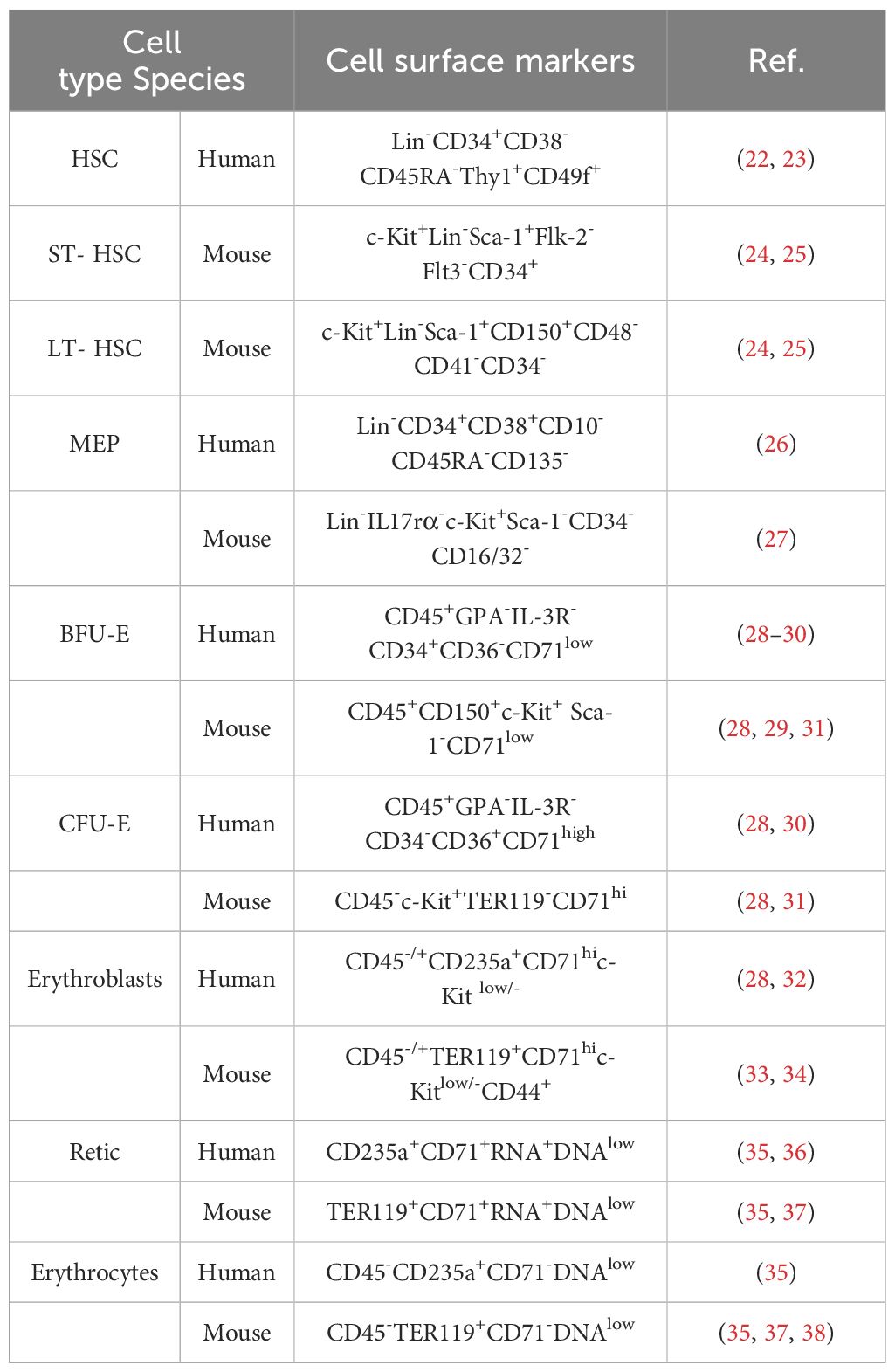
Table 1. Cell markers of the erythropoiesis.
2.2 Molecular regulation of erythropoiesisThe differentiation of HSCs to erythroid cells is regulated by various cytokines and growth factors (Table 2). The first stage of erythropoiesis is regulated by hematopoietic cytokines, such as stem cell factor (SCF; also known as c-Kit ligand), granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), thrombopoietin, IL-11, and transforming growth factor β (TGF-β). Further erythropoiesis is mainly regulated by erythropoietin (EPO), and iron metabolism is essential for hemoglobin synthesis. GATA1, GATA2, KLF1, and TAL1 are key transcription factors involved in erythropoiesis, while the transcription factors, FOG1, and BCL11A, regulate the expression of genes encoding enzymes associated with heme biosynthesis and hemoglobin production (62). Factors involved in the erythropoiesis are listed in Table 2.
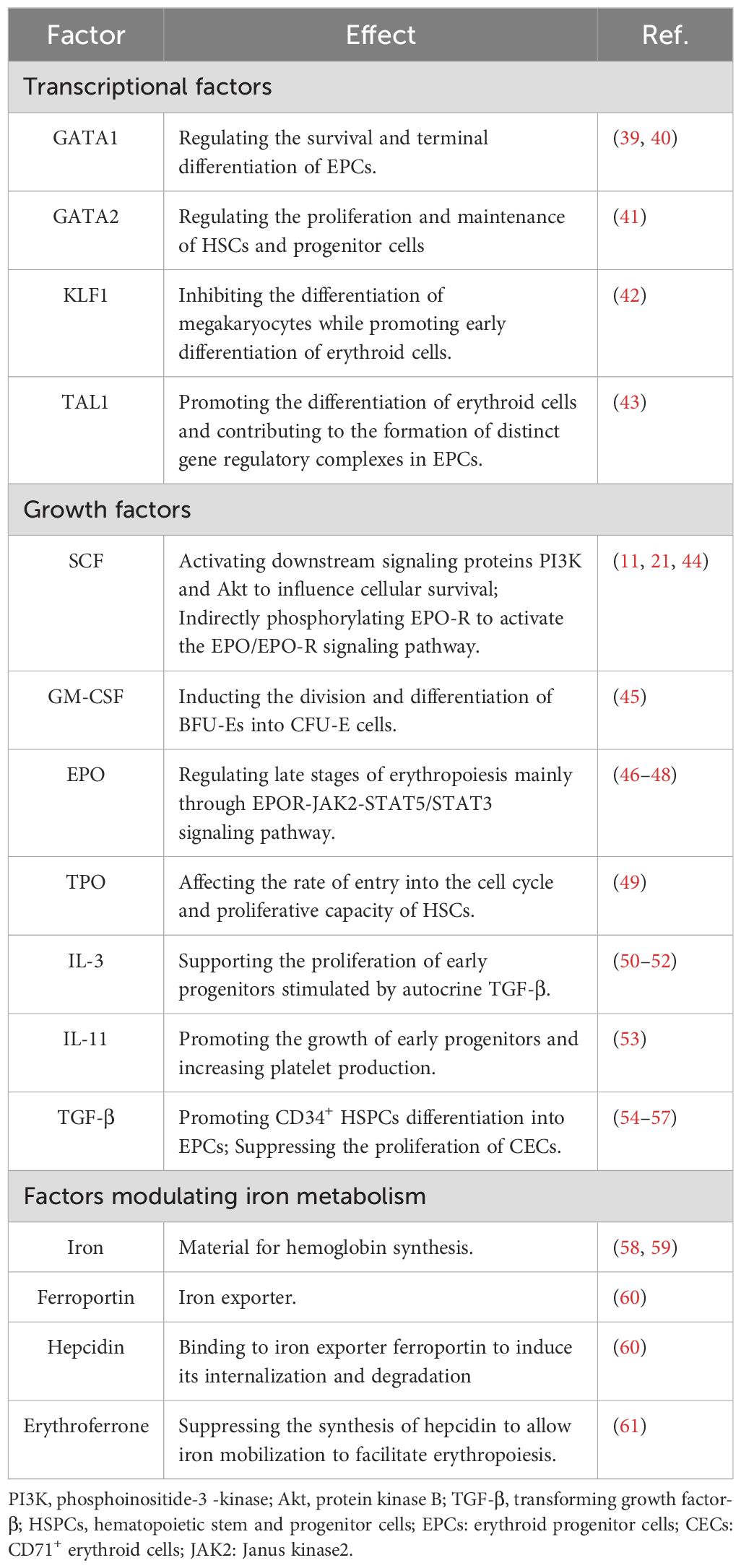
Table 2. Factors involved in the erythropoiesis.
2.3 Macrophages in erythropoiesis and erythrophagocytosis2.3.1 Erythroblast islandsErythroblastic islands (EBI), first discovered by Marcel Bessis in 1958, provide a specialized microenvironment for erythropoiesis (63). EBIs contain a central macrophage surrounded by maturing erythroblasts, and act as the erythroid precursor niche, which supports the bone marrow in producing RBCs at a rate of 2.5 million/second at homeostasis in adult humans (7). Terminal erythroid differentiation occurs within EBIs, where late CFU-Es mature into reticulocytes (64). Macrophages in the EBI secrete growth factors to support erythropoiesis, provide iron for hemoglobin synthesis, phagocytose extruded nuclei, and prevent toxic effects of free DNA (65, 66). Both mouse and human EBI macrophages express EPO-R, while EPO in the niche acts on erythroid cells and EBI macrophages simultaneously, to promote erythropoiesis. Under stress conditions (see section 2.4), RBCs are mainly produced through splenic erythropoiesis, which is distinct from bone marrow steady-state erythropoiesis (67). Impaired EPO-R signaling in splenic niche macrophages significantly inhibits the differentiation of stress erythroid progenitors (68). Further, EBI macrophage dysfunction can lead to specific erythroid hematological disorders (69).
2.3.2 ErythrophagocytosisRBCs have a life span of around 120 days in the circulation. Macrophages have important roles in phagocytosis of aged or injured RBCs and contribute to iron recycling (70). RBC clearance is regulated by so called “eat me” and “don’t eat me” signals. Interaction of CD47 with SIRPα provides the “don’t eat me” signal (71). When RBCs undergo aging, “eat me” signals, such as phosphatidylserine (PS) and band 3, accumulate on their membranes in a process termed eryptosis (72). PS binds to Tim-1, Tim-4, Stabilin-2, or CD300 on macrophages, generating a pro-phagocytic signal, while band 3 interacts with CR-1 and Fc receptors to facilitate phagocytosis (73). PS also binds to platelets and ECs, which triggers pro-thrombotic risk and compromises the microcirculation (72). Enhanced eryptosis is observed in several clinical conditions, including malignancies (72). Tumor cells can directly interact with RBCs via galectin-4, leading to RBC aggregation (74). Together, RBC aggregation and augmented RBC adherence to the vascular wall due to enhanced eryptosis enable circulating tumor cells to stably roll along the vessel wall at a lower flow rate (75).
2.4 Stress erythropoiesisStress erythropoiesis is a stem cell-based tissue regeneration response that occurs in the spleen and fetal liver (76). Anemia or hypoxia accompanied by inflammation, which occur frequently during cancer development (77, 78), chronic infection (79), severe trauma (80), and chronic psychological stress (81, 82), disrupt the homeostasis between erythroid cell production through steady-state erythropoiesis and clearance of senescent or damaged erythroid cells by phagocytes, inducing stress erythropoiesis (79, 83); this process is regulated by bone morphogenetic protein 4 (BMP4), SCF, Hedgehog, EPO, growth-differentiation factor 15 (Gdf15), and glucocorticoids (GCs) (Figure 2) (84). Under homeostatic conditions, low EPO levels support terminal differentiation of only the most EPO-sensitive progenitors, while other erythroid progenitors undergo apoptosis; however, during stress erythropoiesis, increased EPO levels induce massive and rapid terminal differentiation of all erythroid progenitors (56). In addition, BMP4 and Hedgehog signals restrict the transition of short-term-HSCs to EPO-sensitive stress erythroid progenitors. Immature stress-induced erythroid progenitors maintain stem cell properties, including self-renewal, and can be serially transplanted (84–87). Further, BMP4 and SCF are required for the expansion of stress BFU-E spleen cells under hypoxic conditions (88), while Gdf15 regulates murine stress erythroid progenitor proliferation and controls stress erythropoiesis niche development (89). GCs are also essential for immature erythroid cell expansion during stress erythropoiesis, and act by binding and modulating the transcriptional activity of their cognate nuclear receptor, glucocorticoid receptor (GR) (90).
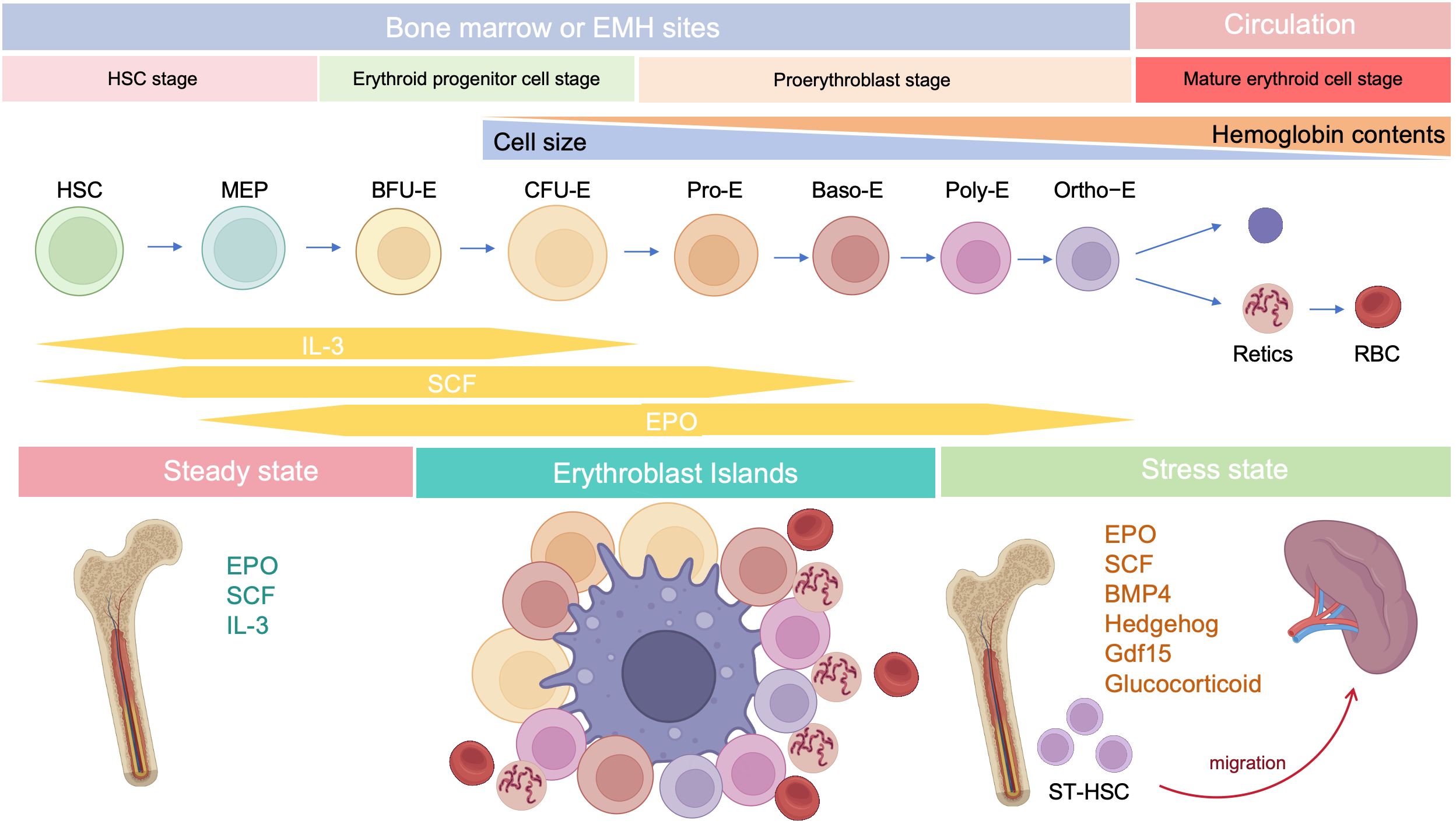
Figure 2. Developmental stages of erythropoiesis after birth. Under steady state, erythropoiesis occurs in the bone marrow, while stress erythropoiesis occurs mainly in the spleen. Erythropoiesis occurs in erythroblastic islands, which contains a central macrophage surrounded by developing erythroid cells.
Although biomarkers of BFU-E erythroid progenitors (Lin-cKit+CD71LowCD150+CD9+Sca-1-) responsive to stress erythropoiesis in the murine spleen are the same as those detected during steady state (91), whole genome transcriptional analysis demonstrated that mouse stress-BFU-E gene signatures are more BMP4-responsive and associated with erythropoiesis and proliferation, relative to those detected in the steady-state (92). Single-cell RNA-seq analysis of human stress-induced erythroid progenitors also revealed a distinct sub-population to that observed under steady-state erythropoiesis (93). Furthermore, splenic BFU-E exhibit different growth properties to their bone marrow counterparts; splenic BFU-E require only EPO to form colonies, while bone marrow BFU-E require EPO and a second factor (94).
Factors upstream of stress erythropoiesis have fundamental immunomodulatory effects. EPO is the principal cytokine regulating erythropoiesis through EPOR; however, EPOR is expressed not only on erythroid cells, but also on immune cells, such as macrophages, dendritic cells (DCs), mast cells, and lymphocytes (40). EPO can bind to EPOR and tissue-protective receptor (TPR, an EPOR/CD131 heterodimer), which are important in tissue protection and immune regulation (95). EPO inhibits the induction of genes encoding proinflammatory factors, such as TNF-α and inducible nitric oxide (NO) synthase (iNOS), in activated macrophages by decreasing NF-κB p65 activation (96). In addition, EPO suppresses DC maturation through the Jak2/STAT-3/SOCS1 pathway (97). Furthermore, EPO directly promotes regulatory T cell (Treg) proliferation, while inhibiting the expansion of conventional T cells via molecular crosstalk with the IL-2 pathway (98). GCs are required for regulation of stress erythroid progenitor expansion (90); however, GR signaling also has potent anti-inflammatory effects (99). Stress erythropoiesis produces more RBCs and CECs, and both populations possess considerable immunomodulatory functions under various conditions (see below for further details).
3 Immunomodulatory effects of RBCsThe link between RBCs and immune function was reported as early as 1953, when Nelson RA Jr. discovered the phenomenon of immune-adherence between erythrocytes and microorganisms, which augments phagocytosis (1). In 1991, RBCs were reported to bind to IL-8 and prevent its release into the blood, thereby limiting leukocyte stimulation (100). Data reported in 1993 demonstrated that RBCs can bind to several chemokine superfamily inflammatory peptides, indicating that RBCs may act as regulators of inflammatory processes (101). Unlike healthy RBCs, RBCs carrying mitochondria (Mito+ RBCs) augment inflammation. Furthermore, oxidative stress and RBC senescence generate a forward feedback cycle, resulting in the release of pro-inflammatory microparticles (MPs), Hb, heme, and iron, and the breakdown products generated by hemolysis have remarkable effects on immunological functions (Figure 3).
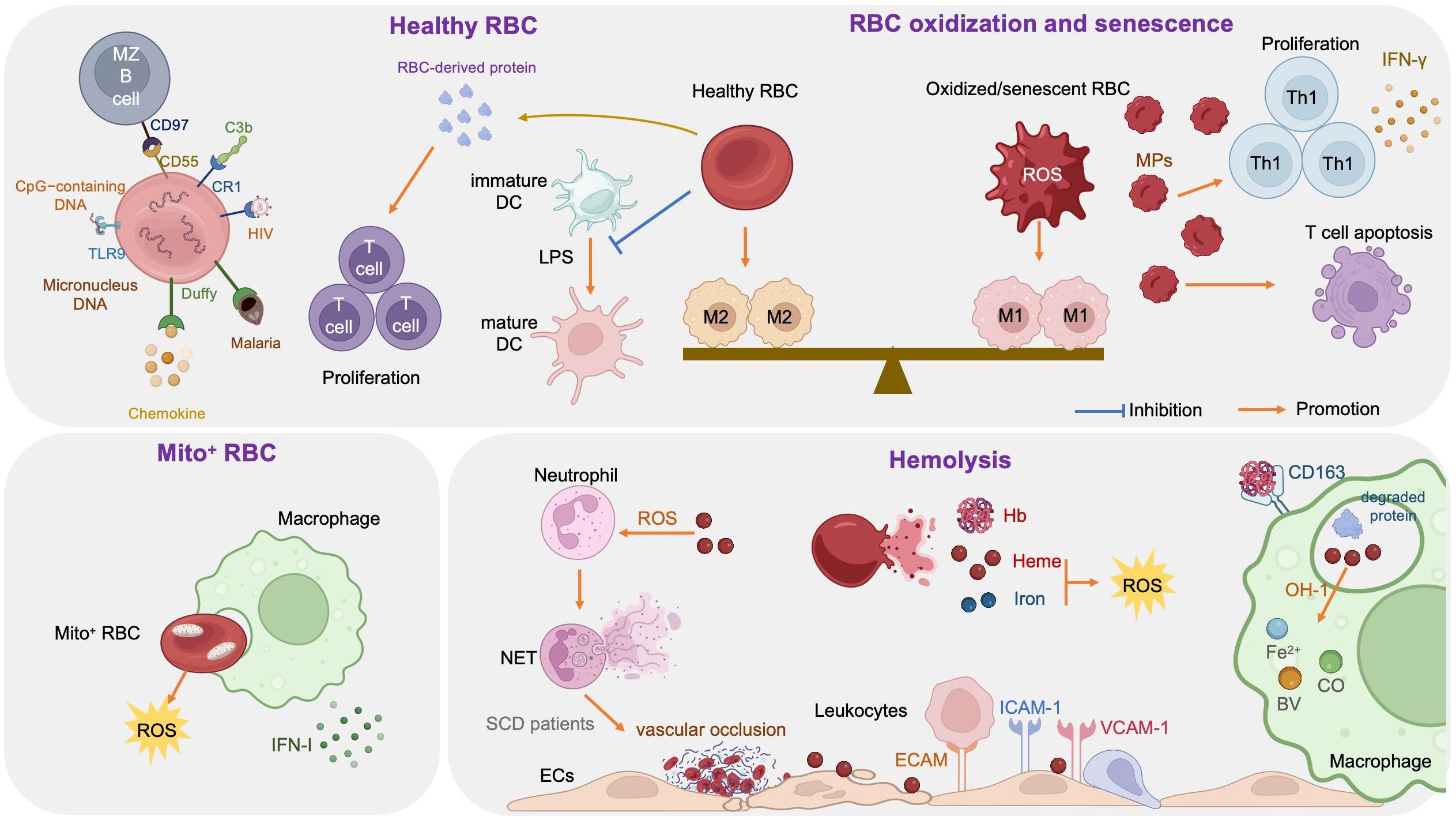
Figure 3. Immune regulation effects of red blood cells (RBCs).
3.1 Healthy RBCs and immune regulationRBCs modulate innate and adaptive immunity mainly through their surface molecules (proteins, lipids, and carbohydrates) and potent antioxidant capacity (102); they express large amounts of the key complement regulators, CD55, CD59, and complement receptor type 1 (CR1, also referred to as CD35), where CD55 inactivates C3 convertases generated by all three complement activation pathways, CD59 prevents membrane attack complex formation by preventing C9 incorporation, and CR1 recognizes collagen-like regions of C1q, mannose-binding lectin, C3b, and C4b, to remove complement-tagged inflammatory particles. For example, in patients with HIV, the virus binds to RBCs via C1q-CR1 interaction (103). Further, CR1 is decreased on RBCs in patients with coronavirus disease 2019 (COVID-19), resulting in consistent inflammatory responses and tissue damage (104, 105).
Toll-like receptor 9 (TLR9), a nucleic acid sensing receptor, is expressed on the surface of mammalian RBCs. Under basal conditions, RBCs bind cell-free mitochondrial DNA (mtDNA) through TLR9 and mediate DNA scavenging to prevent unnecessary inflammation (106). Further, in the context of inflammation, TLR9 binds to CpG-containing DNA derived from bacteria, plasmodia, and mitochondria, which drives innate immune activation and red cell clearance (107). Erythroid-specific TLR9 deletion blocks erythrophagocytosis and decreases local and systemic cytokine production (107). During viral pneumonia and sepsis secondary to COVID-19, RBCs also exhibit protein oxidation, together with decreased antioxidant capacity, increased glycolysis, an altered membrane lipidome, and elevated mtDNA binding, which may contribute to anemia and disease severity (102, 108).
Moreover, RBCs can induce DCs toward an immature/tolerogenic phenotype in response to lipopolysaccharide (LPS), through a CD47-dependent mechanism (109). Mechanosensing by RBCs also ensures exposure of splenic type-2 conventional DCs to blood flow, allowing them to capture circulating antigens, while retaining them in the spleen through CD55-CD97 signaling (110); the same mechanism is also important for marginal B cell retention and function (111). In addition, Duffy blood antigen, primarily expressed on the surface of RBCs, can potently bind to multiple chemokines (112, 113).
Transfusion of fresh RBCs under noninflammatory conditions will reduce RBC clearance and therefore lessen macrophage loading with heme, as well as up-regulating heme oxygenase (HO), shifting macrophages toward the anti-inflammatory M2 state (112). Protein factors released from RBCs, such as Hb and peroxiredoxin II, can sustain normal and leukemic T cell growth and survival (113). RBCs can also synergize with TCR/CD3-mediated activation signals and enhance T cell survival and proliferation through a calcineurin-dependent mechanism (111).
3.2 Mito+ RBCs and immune regulationProgrammed mitochondrial removal occurs during normal erythropoiesis (114). A hypoxia-inducible factor-mediated metabolic switch and consequent activation of the ubiquitin-proteasome system precede, and are necessary for, autophagic mitochondria removal, and disruption of this pathway leads to accumulation of RBCs carrying mitochondria (Mito+ RBCs) (115). This process is defective in patients with systemic lupus erythematosus (SLE) (115) and sickle cell disease (SCD) (116), as well as in aged mtDNA mutator mice (117). In patients with SLE, Mito+ RBC levels are correlated with disease activity, and antibody-mediated Mito+ RBC internalization by macrophages induces type I interferon (IFN-I) production through cGAS/STING activation (115), while Mito+ RBCs may contribute to SCD pathophysiology via high reactive oxygen species (ROS) production (118).
3.3 Oxidized or senescent RBCs and immune regulationRBCs are frequently exposed to various stressful conditions during their lifespan, including oxidative stress, osmotic shock, and mechanical squeezing (119), and consequently accumulate damage which influences their functions. Senescent RBCs show pathologic properties, including decreased deformability (120), MP release (121), increased hemin-carrying Hb (122), and surface antigen modification (123). RBC senescence occurs alongside oxidative stress and in turn becomes a source of ROS, which serves as an important signal of RBC senescence (124). Accumulation of oxidized lipids, such as 4-hydroxynonenal, may induce vascular inflammation (125, 126). At the molecular level, the major features of senescent RBCs are Band 3 clustering or breakdown (127), PS externalization (128), loss of glycophorin A, and reduction of CD47 expression (124). Consequently, senescent RBCs lose the ability to control LPS-induced DC maturation (129).
Oxidized or senescent RBCs or RBC-derived MPs are potential modifiers of T cell responses, which enhance mitogen-driven T cell proliferation and apoptosis through an antigen presenting cell- and cell contact-dependent mechanism, and regulate IFN-γ production from T helper 1 cells (124). Moreover, oxidized RBCs release Hb, heme, and iron which are both sources of radicals and able to activate ECs (130) and innate immune cells, such as monocytes (131), in a proinflammatory manner, as detailed below (see section 3.4). Stored RBCs display senescence-related changes, such as reduced structural integrity, MP release, and iron overload, and the transfusion of stored RBCs exacerbates existing lung inflammation and promotes lung injury, due to loss of Duffy antigen expression and their chemokine scavenging function during storage (132). Further, rapid clearance of transfused stored RBCs by macrophages polarizes the macrophages toward the classical M1 phenotype, with a huge Hb iron load (112, 133). In addition, packed RBCs suppress T cell proliferation via cell-cell contact and inhibit T cell activation via ROS-dependent signaling (134).
3.4 Hemolysis and immune regulationRBCs are highly differentiated cells with an elegant structure that allows them to survive under continuous shear stress when transiting, making them ideal messengers between distant organs. The erythrocyte membrane skeleton is a polygonal 2D lattice structure, consisting of lipids, proteins, and carbohydrates (135, 136). The skeleton attaches to the cell membrane through the spectrin-actin junctional complex (adducin, dematin, and P4.1 interact with band 3, GPC/D, and Glut1) and the ankyrin complex (137). Disorders of the RBC cytoskeleton or dehydration cause hemolytic anemia, which is associated with altered immune regulation, as hemolysis breakdown products, including hemoglobin, heme, and iron, have remarkable effects on immunological functions (138).
3.4.1 HemoglobinHemoglobin (Hb) is an iron-containing protein in RBCs formed from globin and heme (Fe2+ protoporphyrin-IX). When large amounts of Hb are released into the plasma from damaged RBCs, the scavenger protein haptoglobin (Hp) can rapidly bind with cell-free Hb, to generate a Hb-Hp complex, which neutralizes the pro-oxidative effects of Hb (139). When Hp binding capacity is saturated, heme in free Hb is easily oxidized to hemin (Fe3+ protoporphyrin-IX) in the circulation. Free Hb triggers vascular and organ dysfunction through extravascular translocation, NO inactivation, oxidative reactions, hemin release, and activation of downstream signaling pathways (see section 3.4.2) (139). Hp-Hb complexes bind to the CD163 receptor expressed on macrophages and hepatocytes and are subsequently digested, releasing heme into the cytoplasm (140). Free Hb enhances platelet activation by binding to ADP, as well as by abrogating the inhibitory effect of NO (141).
3.4.2 HemeHeme is an important iron-containing porphyrin molecule and with crucial roles in cell protection, apoptosis, inflammation, and immune disorders (142). Hydrophobic hemin intercalates into cell membranes. Hydrogen peroxide from various sources splits the heme ring and releases free redox-active iron, which catalytically amplifies ROS production. Consequently, heme regulates inflammation mainly by acting as a pro-oxidant in macrophages, neutrophils, and ECs (143). Furthermore, heme can selectively bind to receptors, transcription factors, and enzymes (89).
Heme stimulates monocyte differentiation into splenic red pulp macrophages and bone marrow macrophages by promoting degradation of the transcriptional repressor, BACH1, and consequent expression of the transcription factor, SPI-C (144). Heme can also act as a pro-inflammatory second hit in macrophages by aggravating LPS-induced TLR4 signaling, or induce an anti-inflammatory response (M2 macrophages) via induction of SPI-C and HO-1, an inducible isoform of HO (145). Moreover, heme impairs phagocytosis by inhibiting cytoskeleton dynamics through the DOCT8/Cdc42 signaling pathway (146). Heme can also induce Treg expansion in purified T cell-monocyte cocultures by upregulating HO-1 in nonclassical monocytes (138).
Heme promotes neutrophil migration by stimulating macrophage-derived leukotriene B4 (147) and activating protein kinase C and G-protein-coupled receptors in neutrophils (148, 149), which induce chemokine expression and ROS production. During neutrophil development in patients with SCD, heme regulates neutrophil differentiation and can cause defective oxidative burst through HO-1 induction (150). Heme can also inhibit neutrophil apoptosis in vitro through the phosphoinositide 3-kinase (PI3K) and NF-κB pathways (151). Further, heme can induce neutrophil extracellular trap (NET) formation through ROS signaling, to protect the host against infections (152); however, in patients with SCD, NETs can enhance the adherence of erythrocytes and platelets to the endothelium and induce vascular occlusion or lung injury (153).
Free heme interacts with ECs and stimulates the expression of adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1), endothelial cell adhesion molecule (ECAM), vascular cell adhesion molecule 1 (VCAM-1), P-selectin, and others, through heme-mediated ROS and NF-κB signaling pathways (154). Leukocytes attach tightly to endothelium through adhesion molecules and migrate to tissue parenchyma, which promotes vascular occlusion and subsequent tissue ischemia (154, 155). In addition, cell-free heme and heme-loaded microvesicles activate the complement system via the alternative pathway in both serum and on the surface of ECs. Heme also upregulates P selectin, C3aR, and C5aR expression, and downregulates that of CD46, on ECs, which contributes to endothelial damage and vascular occlusion in patients with SCD (156).
3.4.3 IronFree heme is catabolized by HO into three products: biliverdin, carbon monoxide (CO), and Fe2+ (142), where biliverdin is converted to bilirubin, and both CO and bilirubin have potent anti-inflammatory and antioxidant properties, whereas Fe2+ enhances oxidative stress, thereby promoting ferroptosis (157). Fe2+ binds to the iron storage protein, ferritin, which has cytoprotective and anti-oxidative effects, as well as a role in iron storage. Ferritin was first discovered as a suppressor of granulocyte and macrophage production in 1981 (158). Further studies demonstrated that ferritin comprises two functionally distinct subunits: ferritin H and L (159). H-ferritin can suppress T cell proliferation in response to mitogens and impairs B cell maturation (159), as well as helping to mediate the protective effect of HO-1 against oxidative stress (160). Moreover, H-ferritin is a negative regulator of CXC chemokine receptor 4 in receptor-mediated cell migration (161). L-ferritin overexpression in LPS-induced Raw264.7 cells can significantly decrease the production of pro-inflammatory cytokines (TNF-α, IL-1β) and NO and inhibit MAPK and NF-κB activation (162).
4 Immunomodulatory effects of CECsThe term CECs refers to immature erythroid cells, including erythroblasts and reticulocytes, which are physiologically enriched in the spleen and cord blood of neonates, but rare in adult bone marrow (45). CECs are characterized by expression of CD71 and glycoprotein A (CD235a)/glycoprotein A-related protein (Ter119), as CD71+TER119+ cells in mice and CD71+CD235a+ cells in humans. CD71 is also known as transferrin receptor 1 (TfR-1), a type II transmembrane protein important in cellular iron uptake and iron metabolism (163). CD71 is a surface marker for erythroid cells from BFU-E to reticulocytes, which first appears in BFU-E, reaches its highest expression levels in Baso-E and Poly-E cells, then declines in Ortho-E cells, and finally disappears in mature erythrocytes (33).
There are three general CEC subtypes: early-stage CECs, EDMCs, and late-state CECs, each with differing immunosuppressive abilities. Erythropoietic tracking showed that CD45+CD71+TER119+ cells are enriched with stage I–III precursors, while CD45-CD71+TER119+ cells contain more terminally differentiated stage III–V erythroid cells (164). Recent studies have indicated that CECs at the earliest stages are more potent immune response suppressors (164–166). The various surface markers and functional properties of the three CEC subtypes are summarized in Table 3, the immunomodulatory effects of the CECs are summarized in Table 4 and Figure 4, and the immunomodulatory effects of the CECs in diseases are summarized in Table 5.
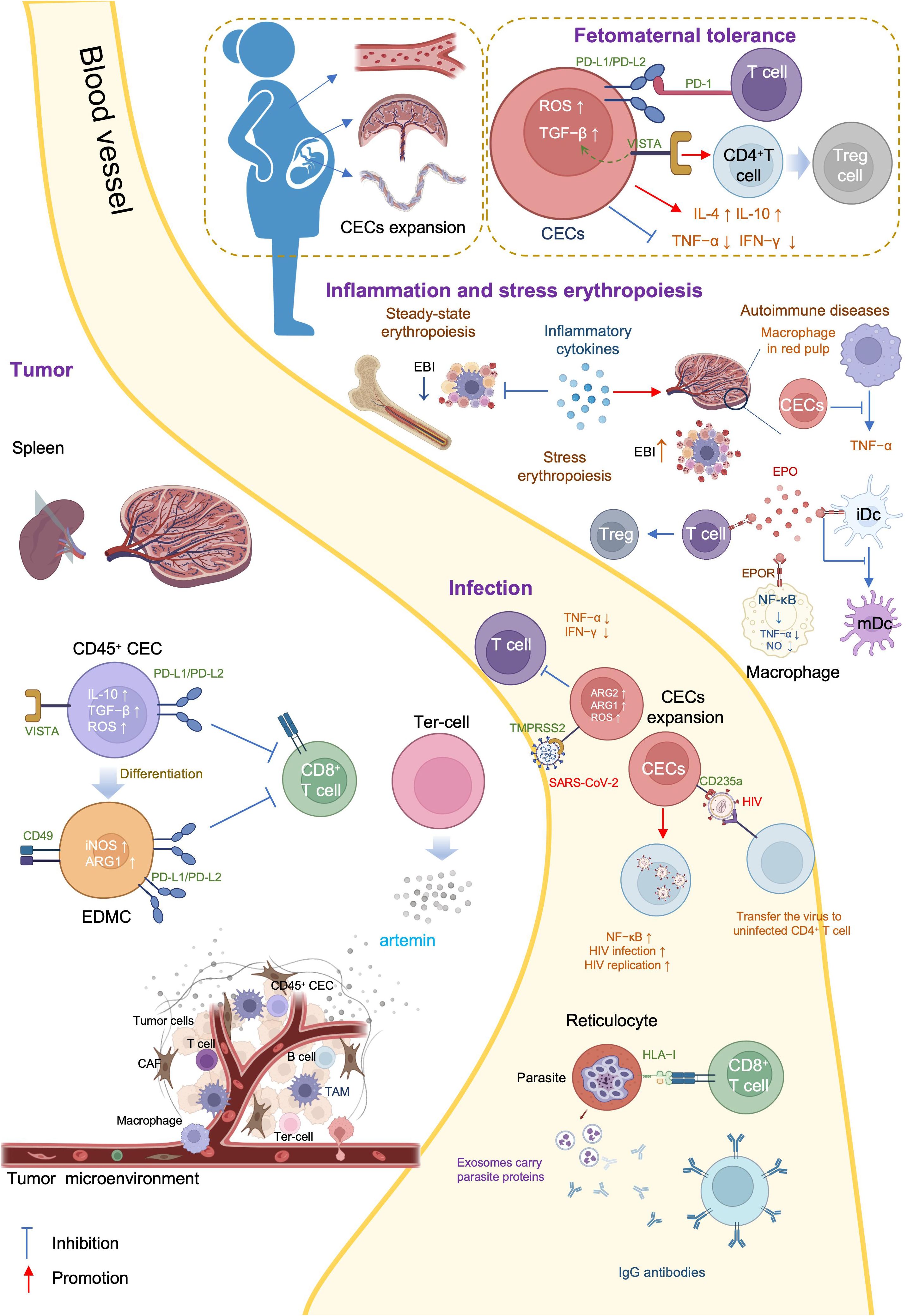
Figure 4. Mechanisms and immunoregulation effects of CECs.
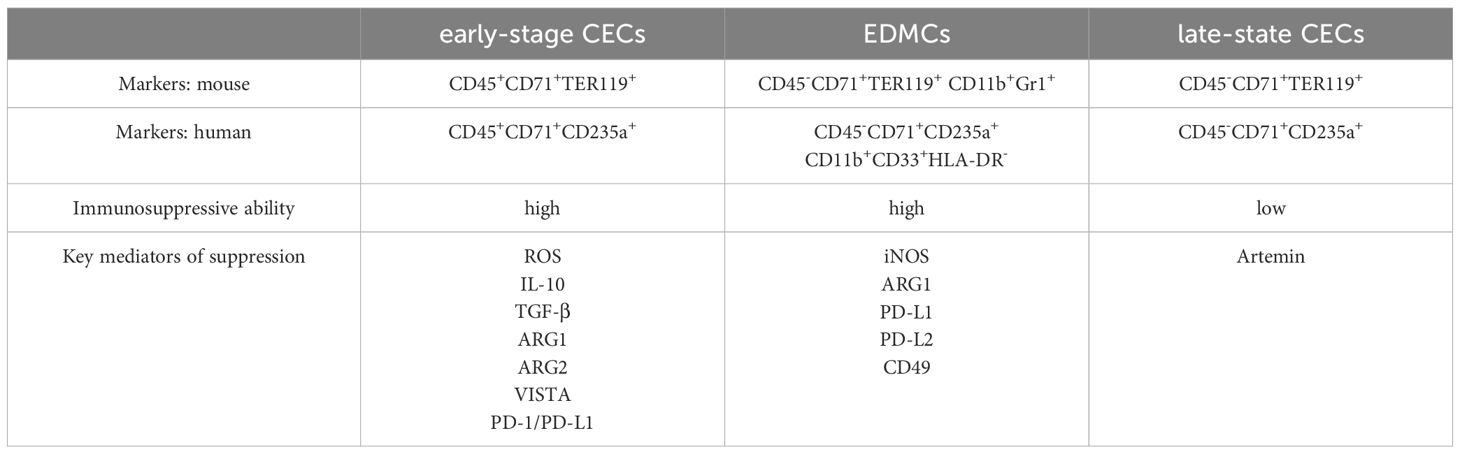
Table 3. Phenotypes of the CECs.
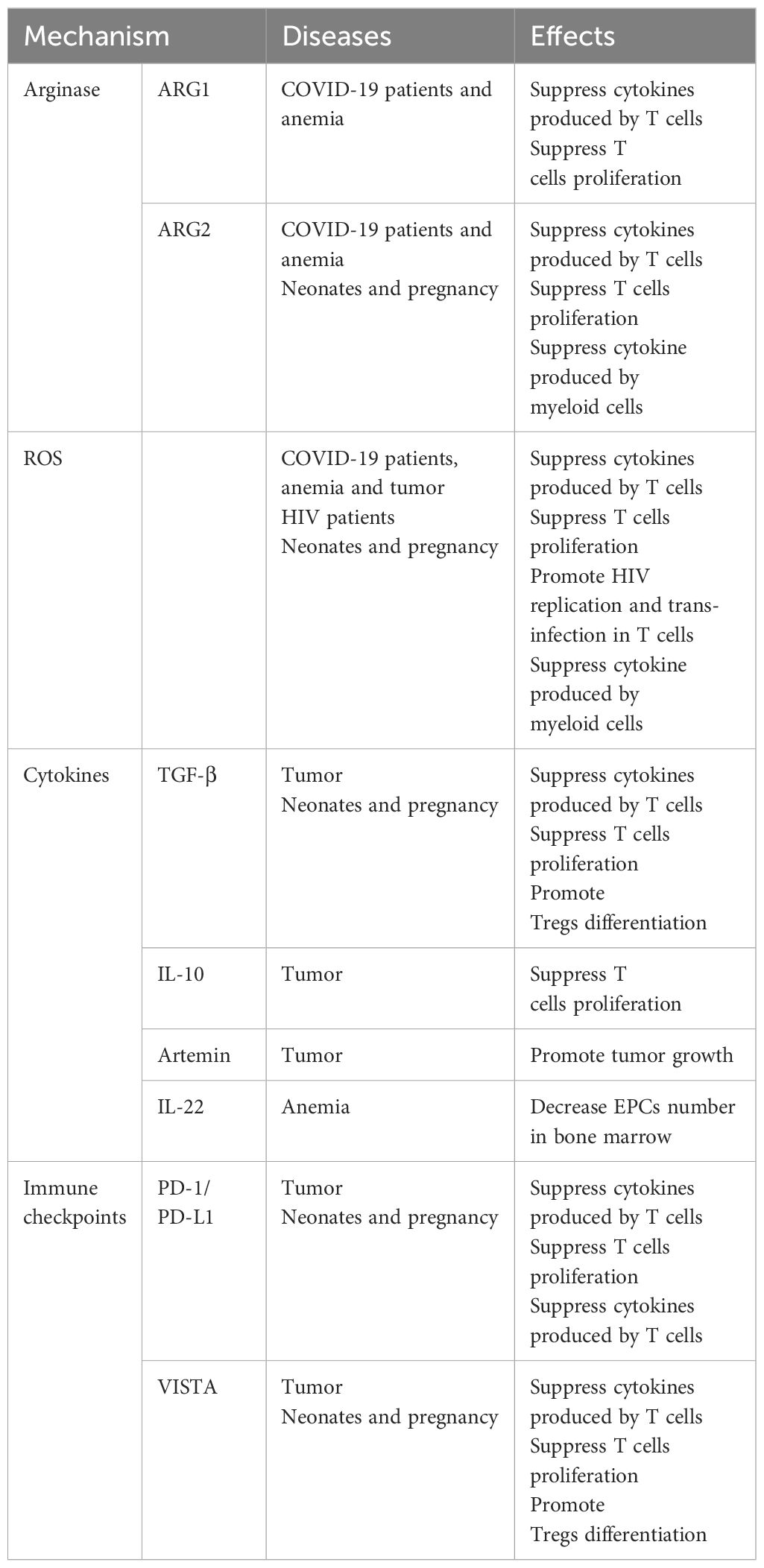
Table 4. Mechanisms of the CECs in immunoregulation.
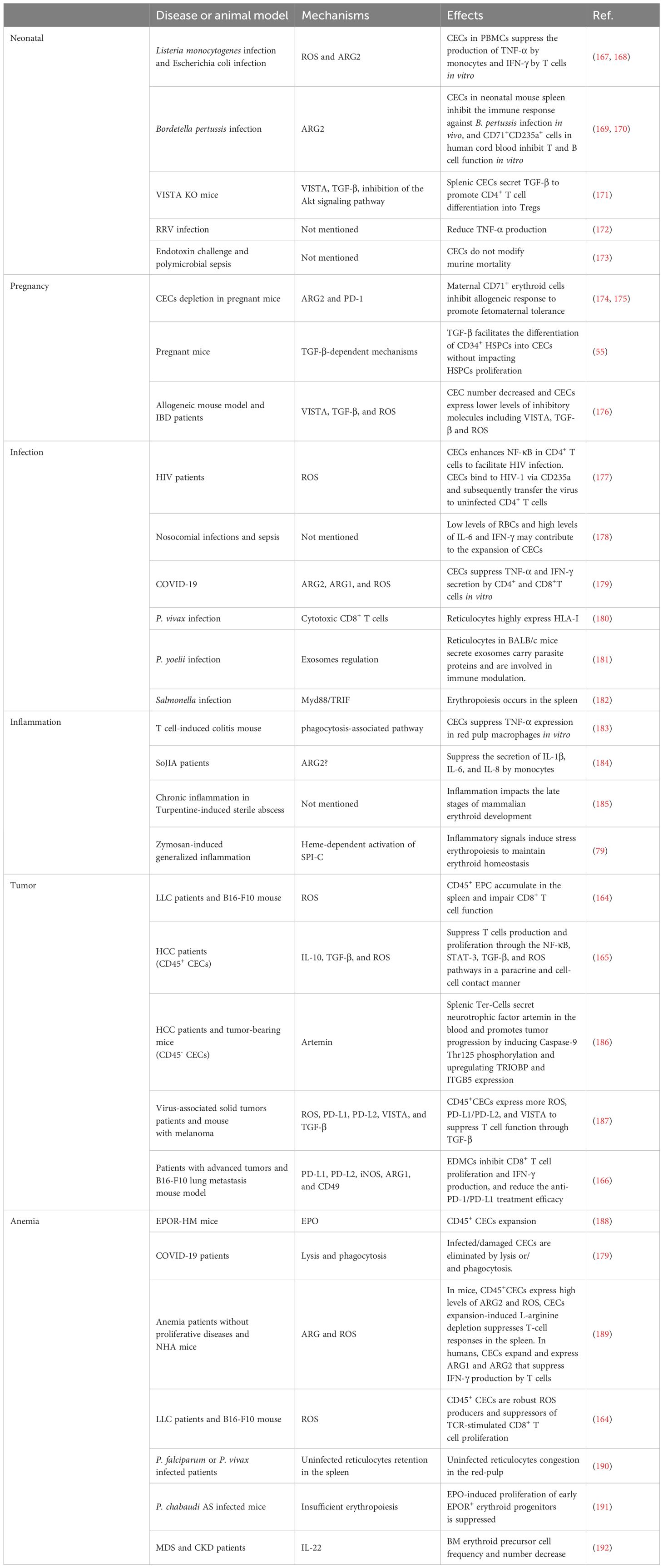
Table 5. Immunomodulatory effects of the CECs in diseases.
4.1 CECs in neonatal and pregnancyErythroid cells play a crucial role in immunological regulation during the neonatal period and in maternal-fetal tolerance. Mouse placental erythroid cells are mainly CD45+ and secrete the chemokines, CCL2, CCL3, CCL4, and CXCL1 (193). Further, CECs are abundant in mouse neonatal spleen and human cord blood, and possess unique immunosuppressive properties (167). CECs are abundant in the liver of children with biliary atresia (BA), and suppress the activation of hepatic mononuclear cells (172). Further, CECs are numerous in the peripheral blood of human newborns, but decline rapidly by 4 weeks of age (168).
CECs influence neonatal infections through various mechanisms. Bordetella pertussis is a common neonatal respiratory tract pathogen and CECs prevent the recruitment of immune cells to the mucosal infection site (167). CECs from human newborn peripheral blood mononuclear cells (PBMCs) suppress TNF-α production by CD14+ monocytes and IFN-γ production by T cells (168). Further, ablation of CECs enhances the innate immune response by increasing the production of protective cytokines, including IL-17, IFN-γ, TNF-α, and IL-12 in B. pertussis-infected lungs (169, 170).
L-arginine is essential for T cell proliferation and function (194). Arginase (ARG) depletes L-arginine, thereby inhibiting T cells, and is encoded by two recently-discovered genes, Arg1 and Arg2 (195). ARG1 is expressed in the cytosol, whereas ARG2 localizes to mitochondria. Neonatal and human cord blood CECs express ARG2 and ablation of CECs augments B. pertussis-specific T cell responses in the lung and spleen on re-infection or vaccination (170). In addition, ablation of CECs also induces enhanced systemic and mucosal B. pertussis-specific antibody responses (170). Accordingly, CECs in human cord blood can suppress T and B cell functions in vitro (170). Regarding innate immunity, CECs inhibit B. pertussis phagocytosis via ARG2 in vitro (169, 170). Such effects of CECs facilitate intestinal colonization with commensal microbes during the neonatal period (167). Depletion of CECs in neonatal mice renders them more resistant to infections by Listeria monocytogenes, Escherichia coli, and B. pertussis, indicating the protective effects against neonatal infectious diseases (167, 168); however, ablation of CD71+ cells failed to modify neonatal mortality in either a model of endotoxin challenge or a model of polymicrobial sepsis (173).
BA is a rare and progressive disease that develops in early infancy (196). Rhesus rotavirus (RRV) infection of neonatal mice induces an obstructive cholangiopathy, which is similar to BA (197). CECs expand in the liver of children with BA or RRV-infected mice and suppress the immune response by reducing TNF-α production. Preemptive depletion of hepatic CD71+ erythroid cells in neonatal mice augments the number of effector lymphocytes and delays RRV infection of the liver and extrahepatic bile duct, suppressing bile duct injury (172). Clearance of CECs before RRV infection renders mice resistant to RRV-induced BA, while repopulation of CD71+ erythroid cells after RRV inoculation promotes long-term survival (172).
CEC-mediated immunosuppression is crucial for fetomaternal tolerance. Both BALB/c and C57BL/6 female mice, and human women are enriched for CECs (198). Further, analysis of 155 umbilical cord blood samples showed that the proportion of CECs was 50-fold higher in cord blood than that in maternal blood (199). Erythropoiesis becomes active during pregnancy, and erythrocytes significantly expand in the peripheral blood (200). TGF-β has an important role in regulating the erythroid lineage differentiation potential of HSCs (201, 202). CECs in pregnant mice express more PD-L1/PD-L2 and suppress T cells expressing programmed cell death protein-1 (PD-1) at the fetomaternal interface (174). Maternal CECs inhibit IFN-γ and TNF-α production to protect the fetus against the allogeneic response. Further, fetal liver CECs also exhibit immunosuppressive activity. A recent transcriptional study demonstrated expression of high levels of galectin-9, galectin-1, and VISTA on the surface of neonatal splenic CECs. CD71+VISTA+ cells produce more TGF-β than CD71+VISTA− cells, and can promote CD4+ T cell differentiation into Tregs (171); however, CECs in human cord blood express negligible amounts of VISTA. Indeed, VISTA expression levels are significantly higher in placental CECs than those in cord blood (171). Thus, both maternal and fetal CECs are essential for fetomaternal tolerance (175). Accordingly, depletion of CECs in pregnant mice induces a proinflammatory immune response, by reducing IL-4 and IL-10 production, while increasing TNF-α and IL-6 levels in placental tissues, which in turn results in fetal resorption (175, 203); however, in pregnant women with inflammatory bowel disease (IBD), CECs are decreased in the peripheral blood, cord blood, and placenta tissue, and express lower levels of inhibitory molecules, including VISTA, TGF-β, and ROS. Accordingly, pregnant women with IBD have lower levels of Tregs and increased immune-activation. Patients with IBD are more likely to have a pro-inflammatory environment in the gastrointestinal tract, which leads to impairment of CECs during pregnancy (176).
4.2 CECs in infectionCECs not only function during neonatal infections, they participate in various infections throughout life.
Acquired immune deficiency syndrome is a systemic disease caused by human immunodeficiency virus (HIV), the genome of which comprises two copies of a 9749 nucleotide sequence packaged in each virion (204). CECs are expanded in the peripheral blood of patients with HIV and there is a positive correlation between CEC frequency and plasma viral load. When cocultured with CD4+ T cells, CECs exacerbate HIV-1 infection/replication, by enhancing NF-κB activation in CD4+ T cells to facilitate HIV infection (177). Meanwhile, CECs bind to HIV-1 via CD235a and subsequently transfer the virus to uninfected CD4+ T cells. Moreover, in the presence of antiretroviral therapy, CECs from HIV-infected individuals contain infective viral particles, which mediate HIV-1 trans-infection of CD4+ T cells (177). CECs are also significantly expanded and possess immunosuppressive properties in the blood of patients with COVID-19. With high levels of ARG2, ARG1, and ROS, CECs mediate immunosuppression by inhibiting CD4+ and CD8+ T cell production of TNF-α and IFN-γ in vitro (179). Furthermore, CD45+ CECs express ACE2, TMPRSS2, CD147, and CD26 and can be infected with SARS-CoV-2 (179).
CECs are also expanded in adult patients with sepsis and serve as predictors of 30-day mortality as well as nosocomial infection development. Low levels of RBCs and high levels of IL-6 and IFN-γ may contribute to the expansion of CECs in sepsis (178). During Salmonella infection, accumulation of CECs in the spleen and increased EPO production are dependent on Myd88/TRIF signaling (182); EPO neutralization reduces the population of CECs in the spleen and slightly improves the host immune response (182).
Malaria is an insect-borne infection caused by the bite of Anopheles mosquitoes, and a major global health problem, with approximately 247 million cases worldwide in 2021 and many more residents of endemic areas having asymptomatic parasitemia (chronic malaria) (205). Different species of malaria parasites exhibit distinct tropism (206). Plasmodium falciparum can invade all stages of erythrocytes while Plasmodium vivax and Plasmodium cynomolgi invade only reticulocytes (207, 208). P. vivax is the most widely distributed human malaria parasite and exhibits a strong preference for immature reticulocytes, with CD71 acting as an anchor receptor (209, 210). Reticulocytes have a more complex and enriched metabolic profile than mature erythrocytes, providing metabolic reservoirs for malaria parasites (206). P. vivax-infected reticulocytes express high levels of human leukocyte antigen class I (HLA-I), which can be specifically detected by cytotoxic CD8+ T cells to protect against intracellular parasites (180). In BALB/c mice, reticulocytes can secrete exosomes when infected by the reticulocyte-tropic non-lethal Plasmodium yoelii 17X strain (211). These reticulocyte-derived exosomes carry parasite proteins and are involved in antigen presentation. Mice immunized using purified exosomes produce IgG antibodies that can recognize P. yoelii-infected RBCs and show increased survival time and altered reticulocyte cell tropism of the parasite (181). Furthermore, during P. vivax infection, parasites invariably affect bone marrow CD71+ cells, inducing dyserythropoiesis and ineffective erythropoiesis (212). Identification and characterization of the reticulocyte receptor, metabolism, and the underlying mechanisms involved in malaria may provide insights to inform the development of novel antimalarial drugs and vaccines.
4.3 CECs in inflammationInflammation is the automatic defense response to tissue injury, and can be classified as acute and chronic, according to its duration. Inflammation modifies bone marrow hematopoiesis towards innate immune effector cells at the expense of lymphoid cells and erythrocytes (79). Inflammatory cytokines, such as TNF-α, limit steady-state erythropoiesis and promote granulopoiesis. Further, mature granulocytes contact the central macrophage of EBIs and alter EBI structures, leading to increased numbers of maturing granulocytes and fewer erythroid precursors (213). In chronic inflammation resulting from sterile abscesses, erythropoiesis is impaired at Ter119+ stages of erythroid development (185). Although inflammation inhibits erythropoiesis in the bone marrow, inflammatory signals induce stress erythropoiesis in the spleen, to maintain erythroid homeostasis. Inflammatory signaling through TLRs enhances erythrophagocytosis by splenic macrophages and augments expression of the transcription factor, SPI-C. In turn, SPI-C couples with TLR signaling to promote the expression of Gdf15 and Bmp4, which encode ligands that initiate the expansion of stress erythroid progenitors in the spleen (79). The spleen is the largest secondary lymphoid organ, and has a wide range of immunologic functions alongside its roles in erythropoiesis, and splenic erythropoiesis alters the histological structure of spleen to become rich in granulomatous lesions and devoid of clear separation between red and white pulp (214).
Autoimmune diseases comprise a range of disorders in which the immune response to self-antigens results in tissue damage or dysfunction (215). In patients with autoimmune diseases, CECs can inhibit inflammatory responses to prevent excessive inflammation and injury. Experimental autoimmune encephalomyelitis (EAE) is an autoimmune disease mainly mediated by specific sensitized CD4+ T cells, which serves as the best experimental model reflecting the autoimmune pathogenesis of human multiple sclerosis (216), and iron-deficient mice fail to develop EAE (217). Management using EPO or its non-erythropoietic derivatives consistently decreases EAE-associated TNF-α, IL-1β, and IL-1Ra production in the spinal cord, and IFN-γ by peripheral lymphocytes, which ameliorates chronic murine EAE (218). IBD inflammation spreads systemically and can cause complications, such as arthritis, cachexia, and anemia. In a CD45-deficient Rag1-deficient mouse model of T cell-induced colitis, an increased number of erythroid progenitors are found in the spleen. These CECs can suppress TNF-α expression in red pulp macrophages in a phagocytosis-dependent manner (183). Further, erythropoiesis-related genes are upregulated in PBMCs of patients with systemic-onset juvenile idiopathic arthritis (SoJIA) (184), while active SoJIA-driven CECs co-cultured with healthy donor monocytes suppress IL-1β, IL-6, and IL-8 secretion. Although ARG2 is the top upregulated gene in SoJIA-driven CECs, cytokine production from monocytes remains suppressed when they are treated using an arginase inhibitor (184).
4.4 CECs in tumorTumors are complex ecosystems, comprising tumor cells and various non-neoplastic cells (219), where non-neoplastic cells in the tumor microenvironment play critical roles in cancer development. Targeting the tumor microenvironment is considered a promising approach for cancer intervention (220). CECs are abundant in both the tumor microenvironment and the circulation and their levels can be used to predict tumor recurrence (165).
Tumor-associated myeloid cells include myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and neutrophils (221), which are important immune cell populations in the tumor microenvironment that are crucial for immune checkpoint blockade efficacy (222). MDSCs can be divided into at least two major subsets: mononuclear MDSCs (M-MDSCs, CD11b+Ly6G-Ly6Chigh) and polymorphonuclear MDSCs (PMN-MDSCs, CD11b+Ly6G+Ly6Clow) (223), where M-MDSCs exert more robust immunosuppression than PMN-MDSCs. Further, erythroid cells can differentiate into myeloid cells in tumors and mediate immunosuppression. Lineage tracking in patients with cancer and tumor-bearing mice revealed that > 30% of erythroid progenitor cells lose their developmental potential and switch to the myeloid lineage, and that these erythroid differentiated myeloid cells (EDMCs) are similar to their myeloid-originated counterparts at the transcription level (166). The phenotypes of EDMCs are CD45+CD235a+CD71+CD11b+CD33+HLA-DR- in patients with cancer and CD45+Ter119+CD71+CD11b+Gr1+ in tumor-bearing mice. EDMCs express more immune inhibitory molecules, including PD-L1, PD-L2, iNOS, ARG1, and CD49, than MDSCs, which may endow EDMCs with the ability to inhibit CD8+ T cell proliferation and IFN-γ production. Accordingly, EDMCs reduce the efficacy of anti-PD-1/PD-L1 treatment (166).
In tumors, CD45+ CECs exert a strong immune suppressive function, mainly by regulating T cells. In Lewis lung cancer, CD45+ CECs are induced by tumor growth-associated extramedullary hematopoiesis (EMH) in the spleen and their transcriptome closely resembles that of MDSCs. As robust immunosuppressors, CD45+ CECs hinder both CD8+ T cell priming in the spleen and effector function in peripheral tissues (164). In hepatocellular carcinoma (HCC) tissues, CD45+ CEC numbers are higher than those of CD45- CECs. Further, CD45+ CECs from patients with HCC inhibit CD4+ T cell proliferation and differentiation and suppress CD8+ T cell proliferation and cytotoxicity by generating factors including ROS, IL-10, and TGF-β (165). In patients with virus-associated solid tumors, substantially greater expansion of CECs occurs in the blood compared with that in healthy controls. CD45+ CECs have more immunosuppressive properties than their CD45- counterparts, mediated by higher levels of ROS, PD-L1/PD-L2, and VISTA. Further, the abundance of CECs in the circulation may be associated with anemia (187). Moreover, CECs in mice with melanoma secrete artemin, while this is not the case for VISTA+ CECs in patients with virus-associated solid tumors (187).
CD45- CECs have lower immunosuppression abilities than their CD45+ counterparts; however, they also play a crucial role in promoting tumor progression. One population of tumor-induced erythroblast-like cells (CD45-Ter119+CD71+, Ter-cells) derived from MEPs (186, 224), accumulate in the spleen of patients with terminal cancer and secret artemin, where artemin is a neurotrophic factor with an important role in cancer progression through its induction of Caspase-9 Thr125 phosphorylation, to maintain cell survival, and upregulation of TRIOBP and ITGB5 expression, to promote invasion. Blocking artemin, or its receptor, GFRα3, signaling inhibits HCC growth in vivo (186). In this context, the phenotype of Ter-cells is CD45-Ter119+ CD71+CD41+CD44+, and they mainly exist in the spleen of advanced-tumor bearing hosts;
留言 (0)