
記住我






While melanin pigments are found in various parts of the body including the skin, hair, eyes, and brain, this article focusses on how differences in skin melanin content may influence drug pharmacokinetics and -dynamics.
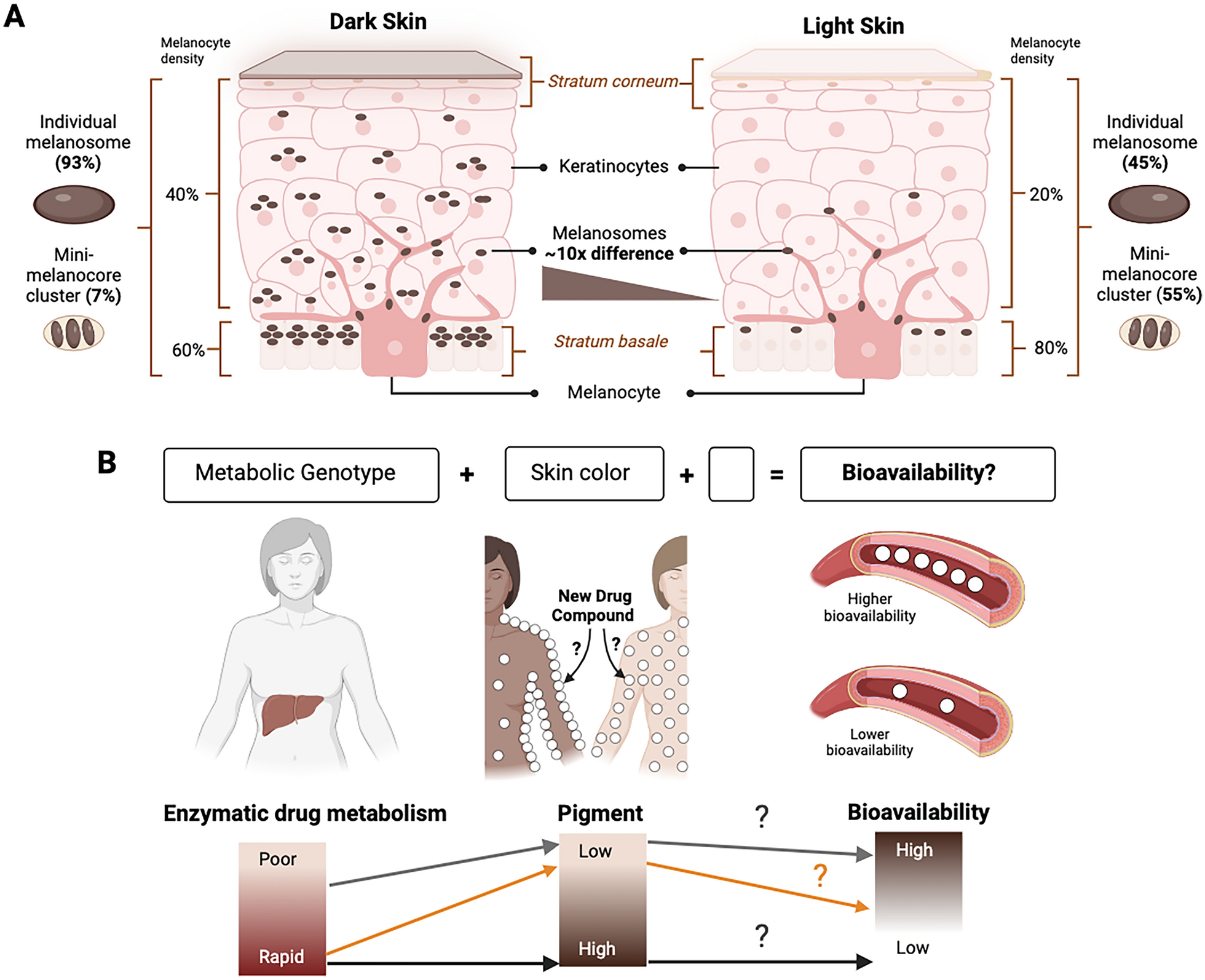
The key cell types contributing to human skin tone are melanocytes, that synthesize melanosomes, and keratinocytes, that store these melanin-containing packages. The quantity and characteristics of melanosomes within skin layers determine variation between light to dark skin tones. Dark skin contains a higher proportion of individual large melanosomes (about 93%), whereas light skin contains higher levels of mini-melanocore clusters (approximately 55%) [13, 14] (Fig. 1A). In light skin, melanosomes are concentrated in the stratum basale, while in dark skin they become distributed more diffusely throughout the layers [13]. The primary difference between light and dark skin, however, is the number of melanosomes present. Dark skin can contain up to ten times more melanosomes compared to light skin [15, 16]. Interestingly, constitutive differences in pigmentation are encoded in the genome as exemplified by an experiment in which fibroblasts isolated from individuals with light and dark skin were reprogrammed into melanocytes. The resulting reprogrammed melanocytes successfully replicated the original human skin phenotypes under the same culture conditions [17]. Indeed, rigorous GWAS demonstrate the contributions of numerous genes to the heritability of skin pigmentation, annotating it as a complex, polygenic trait [8, 9].
Fig. 1
A The skin is composed of multiple layers, from the innermost stratum basale that contains the melanocytes to intermediate layers dominated by keratinocytes and the outermost stratum corneum. The melanocytes produce the melanin-containing melanosomes, which show an approximately 10 times difference in accumulation between individuals with dark and light skin tones. The melanosomes are transferred to the keratinocytes, where they surround the nucleus and protect DNA from UV-induced damage. B This simplified model posits that differential drug responses between human genetic ancestry groups [1] could result from influences of a combination of both the genotype for drug metabolic enzymes and skin eumelanin level of an individual (visible as skin tone) on the bioavailability of drugs. The empty block in the model represents known and/or unknown other variables that may further influence drug bioavailability
Melanin pigments exist in two primary chemical forms: pheomelanin and eumelanin. Both types appear to be present in constant ratios in skins of all tones [15]. However, it is eumelanin that plays a particularly significant role in drug interactions due to its distinctly poly-ionic nature featuring numerous carboxyl and hydroxyl groups. This chemical composition gives eumelanin a high binding affinity for various substances including basic or neutrally charged drugs and metal ions (examples in Table 1).
Table 1 Examples of compounds with binding affinity for eumelaninClinical implications of drug-melanin interactionsThe clinical impact of drug affinity binding to eumelanin has been evaluated most rigorously in retinal toxicity studies. The human eye, particularly the retinal pigment epithelium, contains the highest eumelanin concentration of all tissues in the body. Pharmacokinetic studies focusing on ocular melanin have led to valuable insights: (1) Some eumelanin-associated compounds can impose ocular toxicities such as vision loss, color vision defects, and corneal deposits. These types of drugs include the antimalarial hydroxychloroquine, the antibiotic ciprofloxacin, and the antipsychotic chlorpromazine (reviewed by [20]). (2) For other drugs, local sequestration by eumelanin can be used as a protective mechanism to minimize possibly harmful exposure in systemic tissues [30]. (3) Finally, eumelanin binding has been leveraged for its slow drug release properties to prolong drug action [23, 31]. These mechanisms may be just as relevant for eumelanin in the epidermis and therefore, elucidating the effects of skin pigmentation on the bioavailability of drugs, irrespective of whether they are topically applied or orally administered, could be crucial for addressing safety concerns, optimizing drug delivery mechanisms, and establishing appropriate dosing regimens.
Skin-specific drug-melanin interactions that alter pharmacokineticsThe skin, despite being the largest organ in the body, has been relatively overlooked for potential interactions between eumelanin and drugs or xenobiotics [3]. While the concentration of eumelanin per skin cell may be lower than that found in retinal pigment epithelium cells, the sheer number of cells that constitute skin in a human body contributes to a higher absolute eumelanin content overall. The impacts of human variation in epidermal eumelanin levels on drug pharmacokinetics and -dynamics may be obscured by those of better-studied ancestry-biased genetic polymorphisms in drug metabolic enzymes such as CYPs [2]. However, as we will see below, epidermal eumelanin and drug metabolic enzymes can both potentially contribute to altering the bioavailability of drug compounds (Fig. 1B).
For example, clozapine is an antipsychotic agent primarily indicated for treatment-resistant schizophrenia patients. Its known risk for causing adverse effects necessitates regular plasma concentration monitoring by clinicians after prescribing specific doses. Pardiñas et al. (2023) analyzed clozapine pharmacokinetics in 4,495 individuals from five genetic ancestry groups and found that at the same dose, sub-Saharan African ancestry was associated with lower clozapine concentrations in the plasma compared to European ancestry [32]. GWAS attributed this variation to several genetic polymorphisms linked to the drug metabolic enzymes CYP1A1/1A2 (rs2472297) and UDP-glucuronosyltransferases of the UGT1A family (rs3732218). Polygenic risk scores generated from these genetic loci explained 7.26% of variation in clozapine metabolism.
Coincidentally, the compound clozapine is also known to bind eumelanin [19, 27]. Given that individuals of sub-Saharan African ancestry on average show relatively high levels of epidermal eumelanin [8], the lower plasma concentrations of clozapine observed among individuals of this ancestry could therefore potentially be influenced by clozapine affinity for eumelanin [32] (Fig. 1B).
The effect of eumelanin on the bioavailability of certain drugs is further supported by animal studies: a study in rats led by GlaxoSmithKline (GSK) demonstrated that eumelanin recruited active drug compounds with basic, but not acidic, pH to the skin and eyes [33]. This affected the bioavailability of the basic drugs as demonstrated by increased tissue-to-plasma ratios. Another study performed a time course analysis on the bioavailability of radiocarbon-labelled levofloxacin after 1 hour, 24 hours, and 1 week [22]. Tissues with highly pigmented cells exhibited a significant increase in the tissue-to-serum ratio of the drug, indicating localized drug deposits. Notably, while pigmented hair retained active compounds the longest, pigmented skin and eyes released them slowly over time, extending beyond the serum depletion of the circulating levofloxacin.
Similar animal studies comparing pigmented and albino rats demonstrated that nicotine accumulated to 20 times higher levels in pigmented hair [29]. This contributed to the hypothesis that variation in epidermal eumelanin levels between humans might influence nicotine use and dependence [34]. Links between variation in skin pigmentation and tobacco use were indeed found in a study with individuals of African–American ancestry [35], with a smaller study finding no significant correlation [36]. Furthermore, a follow-up study with individuals of African- and European-American ancestries confirmed an association between smoking cessation and skin pigmentation among males in particular [37]. These observations may have implications for the optimization of nicotine replacement therapies, particularly transdermal delivery systems, depending on individuals’ skin tones. If variation in epidermal eumelanin levels indeed influences nicotine bioavailability, then this would necessitate tailored drug regimens to ensure successful therapeutic outcomes for every patient group.
Despite these examples, differences in skin eumelanin levels may not influence the pharmacodynamics of all compounds that have an affinity for eumelanin. A study on acetaminophen – a drug that binds melanin [19] – found no difference in total plasma levels of acetaminophen between individuals of African- and European-American ancestries [38]. Oxidation clearance of acetaminophen did show ancestry-based differences and was 37% lower in African–versus European-Americans, which could have been partially explained by polymorphisms in CYP2E1 [38].
Taken together, however, and as with all parameters evaluated in preclinical studies that aim to obtain a holistic predictor for the efficacy and safety of a new drug compound, epidermal eumelanin content should be included as a variable just as relevant polymorphisms in drug metabolic enzymes are included as a variable. Inclusion of both will potentiate enhancement of the predictive power of preclinical experiments to generate more robust, generalizable clinical outcomes across patients from diverse racial and ethnic groups and with different skin phenotypes.
INDs and diversityFor INDs, the mandated efficacy and safety testing in the preclinical phase is undergoing significant shifts away from using traditional animal-based models towards using NAMs such as advanced 3D human cell models [39]. Major efforts involving industry, academia, non-governmental organizations, and international regulatory bodies are working collaboratively to benchmark novel tissue-specific NAMs that ensure accurate and reproducible endpoint measurements suitable for IND submissions.
This transition offers a unique opportunity not only to advance towards using human-based in vitro systems for efficacy and safety testing but also to incorporate cell models from a diverse range of human ancestral backgrounds – a feat not achievable with inbred animal models [40]. An inclusive NAM-based strategy would bring much-needed improvements to the translatability of preclinical testing results into clinical research, potentially leading to more accurate predictions of drug efficacy and safety across various demographic groups.
The first CDER/FDA-approved NAMs for IND submissions are 3D skin cell models. These are models for ocular irritation testing with human cornea-like epithelium, dermal irritation testing with 3D reconstructed human skin models, and phototoxicity testing via 3T3 Neutral Red Uptake (NRU) assays.
Of the skin endpoints, the FDA guidelines mention melanin only for phototoxicity testing [41]. The guidelines acknowledge that “Compound binding to tissue components (e.g., melanin, keratin) is one mechanism by which tissue retention and/or accumulation can occur” and propose that a “single-dose tissue distribution study, with animals assessed at multiple timepoints after dosing” will provide an adequate assessment. However, the guidelines also posit that “experience with melanin binding drugs suggests such binding alone does not present a photosafety concern“. This is surprising, not only since existing evidence suggests that certain compounds with high binding affinity for eumelanin can effect phototoxicity [42], but also because there is a notable scarcity of publicly available research articles addressing this scientific question, particularly in the context of diverse skin pigmentation profiles [3].
Here, we propose that NAM-based systems could be a valuable additional endpoint measure in relation to how much skin pigmentation could influence the bioavailability of drug compounds with binding affinity for eumelanin. Given the numerous safety and efficacy analyses that need to be performed for IND submissions, adding an endpoint measure may cause worry that performing more experiments will involve additional time and costs. However, a few strategies can be employed for stratification, potentiating a cost-effective analysis pipeline.
A four-pillar workflow for predicting melanin-based differential drug responses in preclinical researchA cost-effective and translatable approach to predicting potential effects of epidermal eumelanin on the pharmacokinetics and -dynamics of INDs can be constituted by four pillars of which the first three are preclinical: (a) biochemical analysis, (b) inclusive in silico analysis and prediction, (c) inclusive analysis of cellular kinetics using NAMs, and (d) clinical trials (Fig. 2). Each preclinical pillar contributes to identifying the probability that drug-eumelanin interactions may have consequences for drug bioavailability using AI modeling. The last pillar involves testing in humans for validation of the impact predicted in pillars a-c. Integrating clinical data for in vitro-in vivo extrapolation (IVIVE) will confirm and enhance the models' predictive power, enable more accurate clinical endpoint stratification, and improve translational relevance for a diverse group of people over time. We dub this inclusive-IVIVE (i-IVIVE).
Fig. 2
An inclusive NAM for predicting the consequences of drug compound-eumelanin interactions on drug bioavailability. Biochemical analysis: This initial step provides a probability estimate of the binding kinetics of drug compounds towards eumelanin in a simplified and highly controlled environment [11]. Cellular binding kinetics: This phase involves measuring eumelanin-related drug binding and dissociation properties in a cellular context. Metadata about the cell models used is critical input for in silico models and predictions. In silico analysis: This computational (AI) step integrates drug compound properties, eumelanin characteristics, biochemical data, in vitro cellular data (including metadata on the cell models used – Table 2), and curated data from the literature (for example on the effects of enzymatic polymorphisms on drug compound metabolism). The computational models process these diverse inputs to refine predictions. Clinical trials: Data obtained in human testing provides continuous feedback loops between model iterations that can help to improve predictions and promote inclusive in vitro-in vivo extrapolation (i-IVIVE). Bold arrows indicate the weight of the initial data flows
Execution of this workflow in a manner that promotes the inclusivity of drug development requires a consideration of patients’ ancestries in each step:
Biochemical assays Testing of purified eumelanin from various animal species is the most established method in biochemical affinity assays (reviewed by [20]). Importantly, these assays offer a first-line indication of potential drug compound-eumelanin interactions and form a scalable method for compound competition assays [20]. There is no evidence for human ancestry-based differences in the quality of eumelanin synthesized. Therefore, for biochemical affinity assays human ancestry does not have to form a key consideration.
Cellular pharmacokinetics assays In vitro testing of drug compound-eumelanin interactions through multilayered cellular assays provides essential insights into the roles of intricate cell architectural features on drug binding dynamics [43]. For instance, pilocarpine has only moderate binding affinity to isolated eumelanin in biochemical assays, yet exhibits a significant 3-10 times higher affinity for eumelanin when evaluated in ocular cell systems [12, 44]. This discrepancy underscores the necessity of considering features of the entire cellular ecosystem, including dual entry kinetics into the cytoplasm and melanosomes, as critical variables that can be assessed in cell models [45]. Cellular melanin affinity binding is critical in phototoxicity assays, while affinity binding and dissociation assays are crucial to generate inclusive predictors for drug bioavailability.
A key technological advance has been the ability to model the continuous transfer of eumelanin from melanocytes to keratinocytes in an in vitro environment. A range of advanced in vitro 3D skin cell models are available (reviewed by [7, 46] and Table 2). RHE models, such as SkinEthicTM (EpiSkin, France) and MelanoDerm™ (MatTek, USA), accurately recapitulate the morphology, biochemical markers, and lipid composition of native human skin and are available with varying pigmentation levels.
Table 2 Minimal cellular data requirements for inclusive model developmentWhat are critical variables to consider when selecting an appropriate skin cell model? (also summarized in Table 2)Considerations for the genetic backgrounds of cell modelsNo single biomarkers have been identified that are indicative for different levels of skin pigmentation across and within human ancestral groups [8, 9]. Therefore, genetic ancestry of cell models is not a selection criterion when it comes to eumelanin-based traits. Nevertheless, obtaining metadata on the genetic ancestry of all cells used in human skin models is still important and supports the four-pillar workflow (Fig. 2). Specifically, knowing the genetic ancestry of melanocytes will enhance the currently limited understanding of population structure-related variation in drug-eumelanin binding kinetics and may prevent researchers from missing (possibly unknown) genetic contributors that can be uncovered by AI. The genetic ancestries of the cell models used should therefore be captured as metadata (Table 2).
Considerations for determining the pigmentation phenotype of skin cell models in vivo and in vitroFor each cell model, an accurate classification of the corresponding donor pigmentation phenotype requires objective, quantitative measures. While the Fitzpatrick scale has been widely used [47], it is limited by its focus on variations among light skin tones associated with European ancestry and its subjective nature. In contrast, spectrometer-based L*a*b* measurements provide quantitative Individual Typology Angle (ITA) values that were shown to correlate with melanosome quantity in the skin [48].
Melanocytes can lose their ability to synthesize melanosomes when grown in vitro. Certain characteristics of the in vitro environment, including the pH and CO2 levels of culture media, can significantly influence melanosome biology [20] and should be monitored for maintenance of physiologically relevant conditions. Vendors such as EpiSkin provide a turn-key solution by offering media alongside their cell models, promoting reproducibility and inter-laboratory standards.
To validate that an in vitro skin cell model accurately represents the donor's in vivo pigmentation profile it is necessary to quantify eumelanin levels and assess melanosome quality and distribution within different skin layers (Table 2). Both the level and distribution of melanosomes vary distinctly across different skin tones (Fig. 1). AI is especially good at interpreting image data [49] and can be leveraged to standardize image analysis and categorization.
Incorporating these inclusive variables is the first step in the development of diverse in vitro skin models that accurately represent variation in human skin pigmentation (Table 2). The next step would be to leverage these for obtaining novel insights. For example, EpiDerm™ skin models (MatTek) have been previously combined with liver spheroids that actively express drug metabolism enzymes such as CYPs in a multi-organ-on-a-chip model [50]. Such model systems could be employed to study drug pharmacokinetics and bioavailability in the context of human ancestral differences in both skin pigmentation and CYP variants present (Fig. 1B). The genetic ancestries of the melanocytes and hepatocytes, including allelic variants for relevant drug metabolism enzymes, form important metadata to move towards more inclusive preclinical research workflows.
The utilization of commercially sourced in vitro skin models significantly enhances the reproducibility of results compared to in-house constructions. Commercial vendors, leveraging high-throughput production capabilities, can implement standardized procedures and phenotypes while conducting comprehensive quality assurance testing. However, it is imperative that product documentation includes extensive metadata such as ancestry information and quantified pigmentation status for each in vitro model (Table 2).
Computational models Given the provision of high-quality preclinical testing data, AI models can expedite the stratification of further preclinical experiments and clinical trials. However, omitting certain variables from the input data will result in a lack of interpretable information on potential effects of these missing variables for the model output (Table 2). Therefore, the current paucity in cell model metadata [10] constrains the potential of inclusive AI-supported predictions and experiment stratification.
The efficacy of AI lies in its capacity to establish correlations between diverse data types and derive logical inferences. Therefore, underpowered studies that, for example, only utilize two cell models in a single experiment (e.g., one of European descent and one of African descent) are insufficient to demonstrate statistical significance. However, the accumulation of properly annotated additional experiments over time, conducted by various laboratories that may even be addressing different scientific questions, enhances the power of AI models to extrapolate patterns and identify risk predictors. The fewer variables added to the model, the lower its holistic view and predictive power, necessitating a higher number of resource-intensive laboratory experiments. This type of AI-focused data aggregation may empower larger pharmaceutical laboratories that have the resources to standardize metadata collection and integrate experimental data from various departments. For academic laboratories, consistently publishing relevant metadata alongside experimental data as outlined in Table 2 will ultimately enable bulk data aggregation and foster AI model development by specialized researchers [55, 56].
A team led by Roche developed a comprehensive in silico model for predicting the biochemical binding of drug compounds to eumelanin with a 91% accuracy. The basicity, lipophilicity, and aromaticity of compounds were factors that drove binding affinity [11]. Biochemical assays have the advantage of being relatively low-cost and being able to control most input variables. Cellular assays introduce a cornucopia of new variables, from cell characteristics and physiology to varying culture conditions. AI models can help us learn how complex variables in human physiology alter melanin's influences on drug kinetics. Providing AI models with as much metadata as possible will ultimately help to extract the information we seek.
Implementation of AI in biology is hampered by limited data availability. Only through diligent long-term reporting and annotation of cell models and culture variables used (an activity that does not add additional costs) can AI models extrapolate logic and bring valuable insights that augment the insights we would otherwise only obtain through manual experimentation [55,56,57].
留言 (0)