
記住我

Within the genus Listeria, twenty-eight species are recognized; however, only two are considered pathogenic: Listeria ivanovii and Listeria monocytogenes (Raufu et al., 2022; Vázquez-Boland et al., 2001). Both L. ivanovii and L. monocytogenes can cause listeriosis, but the majority of cases are attributed to L. monocytogenes and only a few to L. ivanovii. Although much rarer than those caused by L. monocytogenes and L. ivanovii, L. innocua infections have been reported in humans and ruminants (Favaro et al., 2014; Moura et al., 2019; Perrin et al., 2003; Rocha et al., 2013; Walker et al., 1994). Moura et al. (2019) demonstrated the virulence potential of atypical haemolytic L. innocua strains.
Human listeriosis, primarily caused by the consumption of contaminated food, is a severe illness that can manifest in one of two forms: non-invasive gastrointestinal infection in immunocompetent individuals or invasive listeriosis in risk groups, including pregnant women and newborns, the elderly and immunocompromised individuals (Vázquez-Boland et al., 2001; World Health Organization & Food and Agriculture Organization of the United Nations, 2004). In the invasive form, the pathogen surpasses the blood–brain and placental barriers, resulting in septicaemia, meningitis, spontaneous abortion and stillbirth (Lecuit, 2005). In 2022, the European Union reported 2,738 confirmed cases of listeriosis, which is 50 times fewer cases than the predominant gastrointestinal infection reported in humans, campylobacteriosis. Among the surveyed zoonotic pathogens, L. monocytogenes had the highest rates of hospitalization (96%) and case fatalities (18.1%) (EFSA and ECDC, 2023). These highlights the gravity of this major public health issue in developed nations. In addition to posing a significant public health risk, contamination of foods with this pathogen leads to disruptions in production, distribution, and recalls. As a result, it is receiving considerable attention from the food industry and authorities due to the significant economic losses and food waste involved (Li et al., 2022).
The study of L. monocytogenes bacterial model is of undoubtable importance; however, scientific research cannot be directly performed in humans. The investigation of this foodborne pathogen infection in humans has been mainly through reported clinical cases, epidemiological data, genome analysis and the use of infection models. In addition, L. monocytogenes has relatively low incidence in humans and extended incubation periods can be challenging in listeriosis studies, hindering the identification of causing pathogen and contamination routes (Hoelzer et al., 2012; Vázquez-Boland et al., 2001). Although different methods have evolved to better characterize L. monocytogenes, this species is genetically heterogeneous and different typing methods (discussed below) can be used to subtype this species at different levels. Due to the great variety of typing methods available, comparative analysis between studies can be challenging (Koopmans et al., 2023). Additionally, the study of L. monocytogenes virulence potential can be conducted through different host species used as infection models, however, differences in the selected model, infection dose, incubation time, etc. can be difficult when comparing between studies. Several phenotypic and genotypic tools as well as in vitro and in vivo models have been used to evaluate the uneven virulence potential among distinct strains. Given the diversity of L. monocytogenes studies, it is challenging to define criteria that are universally objective, consistent and applicable. Therefore, in this review we aim to explore and analyse the current methodologies utilized for evaluating differences in the virulence potential among strains of distinct CCs (summarized in Figure 1), giving readers an overview of the available literature.

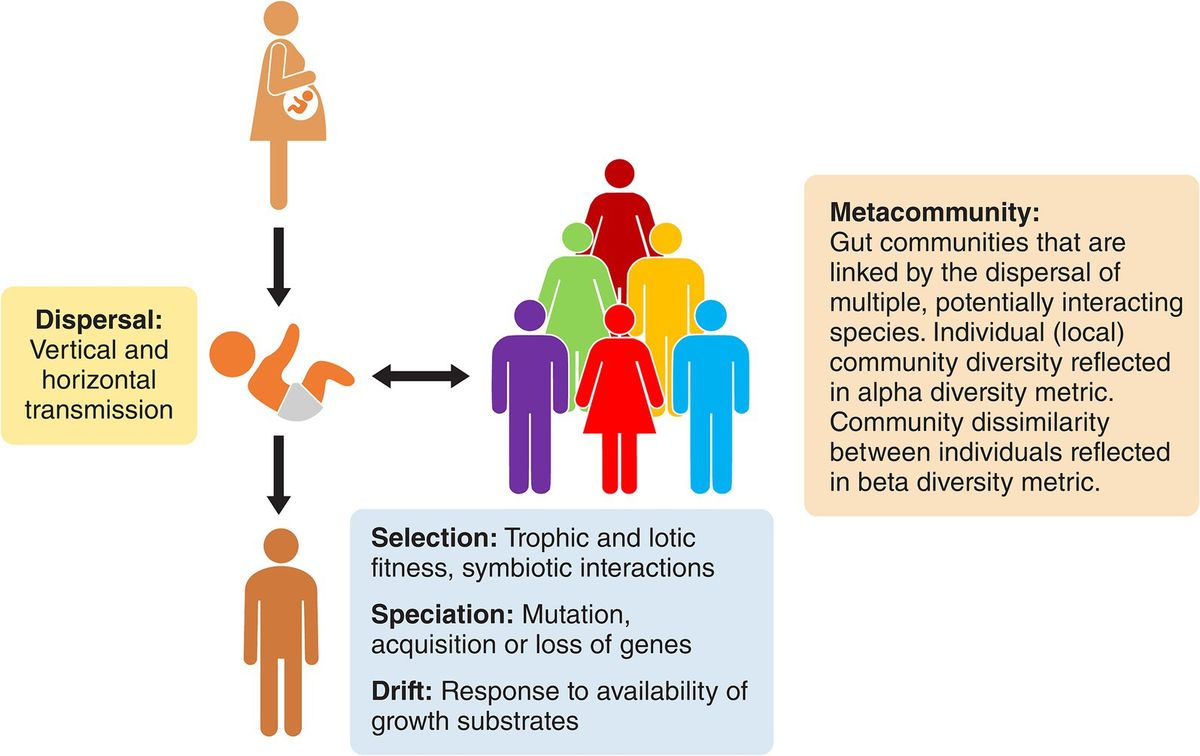
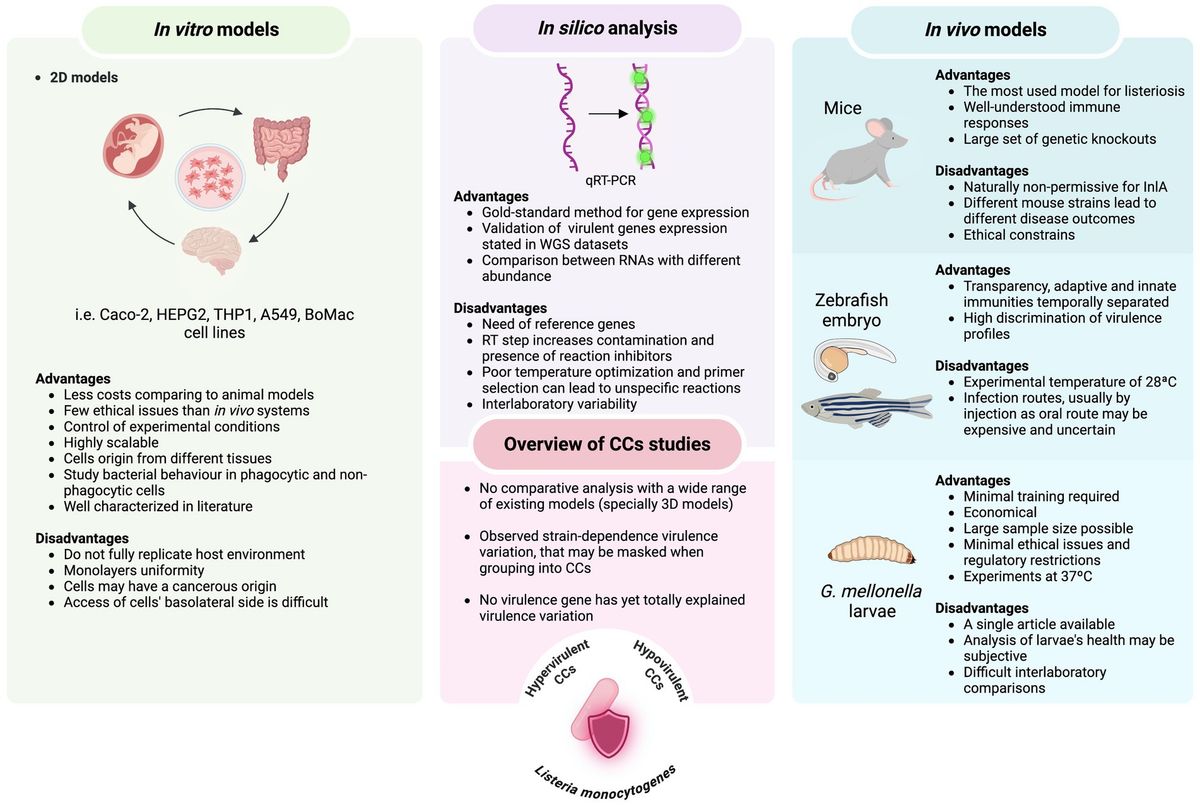
Figure 1. Overview of advantages and disadvantages of infection systems (both in vitro and in vivo models) and molecular approaches used to assess the virulence potential among L. monocytogenes clonal complexes.
2 Typing of Listeria monocytogenesTyping of L. monocytogenes has been essential in epidemiological studies of listeriosis, allowing for the establishment of clonal relatedness among collected isolates. Over the decades, the development and implementation of pheno-and geno-typing methods have made it possible to confirm outbreaks, trace sources of contamination and identify transmission routes within the food chain. Additionally, the increasing adoption of standardized typing methods has facilitated the establishment of effective national and international surveillance systems, enabling the monitoring of evolutionary trends and the generation of comparisons across different geographical regions. This has indubitably had a major influence on the responses and strategies of public health systems worldwide. On the other hand, these methods have massively enhanced our perception of the remarkable biodiversity within L. monocytogenes species and their distribution in different environments. The first method, largely employed in epidemiological studies, was based on the serological antigen structure of the bacterium, specifically on the agglutinating activity of somatic (O) and flagellar (H) antigens (Seeliger and Höhne, 1979; Seeliger and Langer, 1989). This method was gradually replaced by more expeditious methods – namely, a gel-based multiplex-polymerase chain reaction (PCR serogroup) that differentiates, between four major serogroups, including the serovars more frequently isolated from food and patients (> 98%, i.e., 1/2a, 1/2b, 1/2c, and 4b): serogroup IVb (comprising serovars 4b, 4d, 4e), serogroup IIa (comprising serovar 1/2a, 3a); IIb (comprising serovars 1/2b, 3b, 7); and serogroup IIc (comprising serovars 1/2c, 3c) (Doumith et al., 2004). Later, a real-time triplex-PCR assay that differentiates these groups was made available (Vitullo et al., 2013). Although many L. monocytogenes serotypes have been discovered, three major serovars (1/2a, 1/2b, and 4b) are responsible for a substantial fraction of listeriosis cases (about 90 to 95% of human infections) (Schiavano et al., 2022).
Several typing methods have been used for multiple purposes, with genotypic methods being particularly highlighted due to their higher discriminatory power (e.g., amplified fragment length polymorphism (AFLP), multilocus variable-number tandem repeat analysis (MLVA) or ribotyping) compared to phenotypic methods. In the specific case of epidemiological studies, pulsed-field gel electrophoresis (PFGE), based on the analysis of DNA restriction patterns, has been considered the “gold standard” technique for typing L. monocytogenes for many years (Graves and Swaminathan, 2001). However, PFGE has some drawbacks, such as the difficulty of standardizing the analysis of fingerprints, which poses a challenge for inter-laboratory and inter-country comparisons. In addition, while it is valuable for assessing genetic relatedness between isolates, pinpointing sources of contamination and identifying outbreaks, it is not sufficient for establishing comprehensive phylogenetic relationships between strains. Sequence-based typing methods such as multilocus sequence typing (MLST) or multi-virulence-locus sequence typing (MLVST) are more appropriate for this purpose (Maiden et al., 1998; Salcedo et al., 2003; Zhang et al., 2004). Currently, MLST is widely used as a reference method to categorize the clonal structure of bacterial species and to define clonal complexes (CCs) of genetically related isolates, i.e., those descended from the same ancestor. In L. monocytogenes, MLST is based on the sequencing of seven housekeeping genes (acbZ, bglA, cat, dapE, dat, ldh, and lhkA), that allow the determination of sequence types (STs) (Bergholz et al., 2018; Orsi et al., 2011; Ragon et al., 2008). Additionally, Ragon et al. (2008) grouped these STs within CCs, with strains sharing at least six out of seven MLST alleles being assigned to the same CC. Currently, the preferred method for epidemiological and phylogenetic studies has shifted to whole genome sequencing (WGS), which has become more accessible to a broader range of laboratories due to technological advances and reduced costs (Gerner-Smidt et al., 2019). Whole genome sequencing of Listeria provides high-resolution data that not only allows phylogenetic relationships between strains to be determined, but also provides in-depth knowledge of the genomic structure of a given strain, including information on specific virulence factors and other genes that contribute to pathogenesis, as well as potential antibiotic resistance prediction (Hurley et al., 2019; Moura et al., 2024).
This species presents a diverse genetic pool and its virulence potential is very heterogeneous, resulting in an uneven capacity of strains to cause disease (Pyz-Łukasik et al., 2022). Currently, this species is divided into four major evolutionary lineages (I-IV), comparable to subspecies (Liu, 2006; Orsi et al., 2008; Rasmussen et al., 1995; Roberts et al., 2006; Ward et al., 2008; Wiedmann et al., 1997); lineage I includes serotypes 1/2b, 3b, 4b, 4d, and 4e and 7, and is significantly overrepresented in human listeriosis cases (Gray et al., 2004; Orsi et al., 2011); lineage II includes serotypes 1/2a, 1/2c, 3a, and 3c, prevalent among isolates from environmental samples, food, and animal listeriosis cases (Nightingale et al., 2005b; Sauders et al., 2006), and contribute significantly to sporadic cases of human listeriosis (Jeffers et al., 2001); and, lineage III and IV include serotypes 4a, 4c and atypical serotype 4b isolates, which are rare and are mainly associated with listeriosis in animals (Liu, 2006). Clonal complexes are grouped within lineages, for example, CC1, CC2, CC4 and CC6 (serotype 4b, lineage I) and CC121 and CC9 (serotypes 1/2a and 1/2c, respectively, lineage II) (Maury et al., 2016). A methodology for cloning L. monocytogenes and assessing potential human infectivity has been patented (WO2017009198A1).
More than one hundred CCs have been reported globally. The predominance of particular CCs is highly heterogeneous among different sources and regions. In 2011, Chenal-Francisque et al. (2011) characterized the genotypic profile of three hundred isolates collected from 42 countries on five distinct continents, and these isolates were distributed within 111 STs, assembled into only 17 CCs. This reinforces the idea that there is an irregular geographical distribution, with a few prevailing CCs (Chenal-Francisque et al., 2011; Wagner et al., 2022). However, these isolates were collected between 1933 and 2007, and it has been established that the distribution of CCs tends to change over time with some CCs, such as CC9, CC121, CC5, and CC6, emerging more recently (Bergholz et al., 2018). Information on STs/CCs associated with listeriosis outbreaks in European countries during the last decade are presented in Table 1. When molecular characterization of outbreak strains was not available from publications, additional information was collected from the Institut Pasteur MLST database. CC6 and CC8 were the two main CCs, accounting for 17.4% of the total of 23 outbreaks identified. Many studies across European countries have also reported that some clonal complexes, such as CC1, CC2, CC3, CC4, CC5, CC6, CC8, CC9, CC37, CC121, and CC388 are globally prevalent with some geographical disparities (Domínguez et al., 2023; Félix et al., 2022; Maury et al., 2016; Painset et al., 2019). To better characterize this heterogeneity between strains from different CCs, two independent terms have been established: CCs with a high frequency in human clinical cases are considered hypervirulent; conversely, CCs associated with food, persistence in food manufacturing environments, and with a lower frequency in human listerioses cases are considered hypovirulent (Maury et al., 2016). Therefore, CC1, CC2, CC4, and CC6 (lineage I) are considered to be hypervirulent clones since they are clinically related and mainly infect individuals with low or no comorbidities. Contrarily, strains belonging to CC9 and CC121 (lineage II), recognized as hypovirulent clones, are regularly isolated from food and food processing environments. The latter are often associated with individuals with a compromised immune system (Maury et al., 2016). There is also an intermediate classification for those clones that may be in transition from their host-associated lifestyle due to loss of virulence and acquisition of stress resistance genes (FAO and WHO, 2022). In 2018, Fritsch and co-workers also established three different levels of virulence among CCs and STs for risk characterization: hypovirulence, medium virulence, and hypervirulence. The latter, includes, in addition to the previously mentioned hypervirulent CCs, CC224, ST54, CC101 + 90, ST87, ST451, ST504, CC220, ST388, and CC207 (Fritsch et al., 2018). Hypovirulent CCs include CC9 and CC121, as well as CC11, CC19, CC31, CC193, CC199, CC204, and ST124.

Table 1. Reported listeriosis outbreaks in Europe for the last decade.
It is important to note that, although hypovirulent CCs such as CC9 and CC121 are mainly associated with food and food processing environments, cases of invasive listeriosis caused by these CCs have also been reported. For instance, CC121 was considered the second most common CC isolated from human clinical cases in Norway and in France (Fagerlund et al., 2022; Maury et al., 2016).
Despite the potential ability to predict the risk of a specific strain of L. monocytogenes causing disease after consumption of contaminated food, most regulatory authorities worldwide take action when any L. monocytogenes is found in ready-to-eat (RTE) food that is capable of supporting growth, regardless of its strain characteristics. This approach is recommended by the Food and Agriculture Organization (FAO) and the World Health Organization (WHO) (2022), although in some countries risk managers are permitted to use information on L. monocytogenes subtypes to guide risk management decisions. However, the FAO and the WHO encourage the search for other virulence markers to predict, based on genetic virulence profiles (CCs characterization) (FAO and WHO, 2022). The discovery of one or multiple biomarkers that would allow to predict the real virulence potential of a given strain, and a clear distinction between hypo-and hypervirulence would be of great value to reassess the risks associated with different L. monocytogenes strains and to develop appropriate policies that neither overstate nor underestimate the risk posed by each strain. Ultimately, this finding would also contribute massively to the reduction of costs associated with the recall and destruction of contaminated food products and to reduced food waste and its social and economic consequences.
3 Putative virulence biomarkers (core and accessory genome)The L. monocytogenes infection cycle comprises various steps: adhesion and invasion, lysis and escape from the vacuole, cytosolic multiplication, actin-tails polymerization, spread to neighbouring cells, and rupture of a double-membrane vacuole (Luque-Sastre et al., 2018; Pizarro-Cerdá et al., 2012). Some virulence genes are important for infection, such as, InlA-E-cadherin and/or InlB-C-Met (L. monocytogenes internalins-host receptors) for invasion, listeriolysin O (LLO) and phospholipases A and B (PlcA and PlcB) for both primary and double-layer vacuoles disruption, ActA for actin tail polymerization and intracellular motility (Quereda et al., 2021; Radoshevich and Cossart, 2018).
Considering all the above information, a detailed investigation regarding the putative virulence markers linked to both hyper-and hypovirulence is still ongoing, and some interesting findings have been reported. Regarding the core genome, inlA is normally present and expressed as a full-length form within clinical isolates (Lecuit et al., 2001). Premature stop codons mutations (PMSCs) have been found in the inlA gene, resulting in a truncated non-functional internalin in food isolates. In some studies, these PMSCs have been found amid strains from hypovirulent CCs, such as CC9 and CC121, and thus it is hypothesized that in some way, the lower virulence potential of these strains can be justified by the InlA truncation, leading to a reduced capacity to cross the intestinal barrier (Jacquet et al., 2004; Lachtara et al., 2022; Moura et al., 2016). The significant role of InlA-mediated crossing of L. monocytogenes through the intestinal barrier has been described. However, some studies have showed that the inoculation of ΔinlA mutants still resulted in L. monocytogenes infection (Bierne et al., 2002; Bou Ghanem et al., 2012; Disson et al., 2008). Therefore, it was hypothesized that L. monocytogenes employes alternative routes to cross the intestinal barrier. Besides the M cell-mediated translocation in Peyer’s patches, Drolia et al. (2018, 2024) showed that the linkage between Listeria adhesion protein (LAP) and its surface receptor Hsp60 promotes cell disruption by using the cell innate system, consequently leading to bacterial translocation (Drolia et al., 2024; Drolia et al., 2018). These studies have shown that L. monocytogenes can cross the intestine through InlA-independent routes, which could explain the isolation of strains belonging to hypovirulent CCs (normally associated with the production of truncated inlA) in clinical cases. However, to our knowledge no comparative studies have investigated LAP or other InlA-independent invasion factors as putative candidate to distinguish hyper-or hypovirulent strains of L. monocytogenes.
All strains of L. monocytogenes carry the Listeria Pathogenicity Island 1 (LIPI-1), which clusters several fundamental genes for L. monocytogenes pathogenicity (Moura et al., 2016; Vázquez-Boland et al., 2001). These include the hly gene, which encodes a hemolysin – LLO – that provides the capacity to lyse erythrocytes. As mentioned above, this toxin can form a pore and allow the bacteria to escape from the internalization vacuole; thus, this virulence factor is detrimental to the virulence of L. monocytogenes. Another important virulence factor is PrfA, known as the main regulator of virulence genes in L. monocytogenes, such as the prfA, actA and hly genes. However, some studies have reported the existence of non-hemolytic L. monocytogenes strains, belonging to both lineages I and II, that have mutations in either the prfA or hly genes, and consequently a lower virulence potential (Maury et al., 2017).
Regarding the accessory genome, the pathogenicity island LIPI-3 carries eight genes. Listeriolysin S (LLS) encoded by llsA, functions as a bacteriocin with the capacity to modify the composition of the intestinal microbiota by eliminating or hindering the growth of neighbouring bacteria. This virulence cluster is often present within lineage I isolates, especially those from CC1, CC2 and CC6 – constituting a potential marker of hypervirulence (Cotter et al., 2008; Moura et al., 2016; Quereda et al., 2016). Additionally, Maury et al. (2016) identified a novel virulence cluster termed LIPI-4, which aggregates six genes that encode a cellobiose family phosphotransferase system (PTS). This gene cluster is strongly associated with strains of CC4, which are highly relevant to human brain and placental infections (Maury et al., 2016; Moura et al., 2016). Furthermore, it was thought that this pathogenic island was exclusively related to CC4 strains, but isolates from CC87 in China also displayed this locus (Wang et al., 2019; Zhang et al., 2020). These findings suggest that this could be a putative marker of hypervirulence, although it was found that this island was also present in L. innocua – a non-pathogenic species – and thus its role in hypervirulence is still controversial, reinforcing the need for further studies. Another intriguing gene is lmo2776, which acts as a bacteriocin and plays an important role in modulating the intestinal microbiome, mainly targeting Prevotella copri – a common gut commensal that has the capacity to modify the intestinal mucus layer and potentially intensify gut infection. The critical aspect is its significant presence in lineage I strains compared to its low frequency in lineage II strains. Curiously, deletion of lmo2776 resulted in a better spread of the bacteria to the liver and spleen – the primary target organs of L. monocytogenes after crossing the intestinal barrier. This can be explained by the capacity of L. monocytogenes to discriminate between P. copri, preventing exorbitant inflammation and leading to longer periods of infection (Rolhion et al., 2019).
4 Models to study Listeria monocytogenes clonal complexes 4.1 In vivo infection modelsIn virulence studies, both pathogen characteristics and host physiology and anatomy must be considered, as microbial infections result from interactions between pathogens, hosts and the surrounding environment (Prescott, 2022). Preclinical trials using in vivo and in vitro biological models, have provided valuable insights into host-pathogen interactions (Anju et al., 2020; Khan et al., 2018). In vivo systems, used for various purposes from drug development to investigating physiological processes, complement in vitro studies by providing a more comprehensive understanding of biological responses. However, neither system alone is sufficient to make absolute predictions (Khan et al., 2018). Some of these models will be detailed in the following sections (Table 2).

Table 2. Infection models used to study virulence potential among L. monocytogenes CCs.
In order to improve human health research, both mammalian and non-mammalian models are used due to ethical constraints with experiments involving humans (World Medical Association, 2013). The broad host range of L. monocytogenes allows the use of various animal models, such as Drosophila melanogaster (fly), Galleria mellonella (moth), Caenorhabditis elegans (nematode), Mus musculus (mouse), Cavia porcellus (guinea pig), Oryctolagus cuniculus (rabbit), among others (Anju et al., 2020; Prescott, 2022) – some of which will be discussed further. Animal models offer advantages that make them invaluable for human health research: they have identical biological processes, anatomical similarities (especially in vertebrates animals) – which are difficult to replicate in in vitro systems – compatible diseases such as cancer and diabetes, short life cycle and some can be easily genetically transformed to acquire some fundamental characteristics to express the disease phenotype (Kiani et al., 2022). Additionally, in vivo models are essential because they possess some unique characteristics when compared to in vitro models, for instance, the immunity associated with commensals and the intestinal mucosa throughout infection (Eng and Pearson, 2021). Depending on the final objective of the study, several aspects must be considered when selecting the ideal animal model: (1) the pathogen should have a similar tissue and cell affinity as in humans; (2) it should reveal the identical observable disease outcome and immunopathological harm; and (3) it should be susceptible to genetic manipulation (Lecuit, 2007). In addition to animal features, to study L. monocytogenes virulence, understanding listeriosis pathophysiology is crucial to select the adequate animal model. As already mentioned, L. monocytogenes can cross the intestinal, blood–brain and placental barriers. Therefore, pregnant, non-pregnant and geriatric animal models have been used in the study of L. monocytogenes pathogenesis, this was exhaustively described by Hoelzer et al. (2012). Animal models have played crucial roles in the characterization the virulence of L. monocytogenes. Generally, insightful data about the different pathways of bacterial translocation through host’s defensive barriers, the exploitation of host’s immunity to improve disease, performance of dose-dependent assays, the complex host immune responses to infection, the species specificity, virulence factors and strains virulence potential have been emerged from animal models studies (Hoelzer et al., 2012; Koopmans et al., 2023; Lecuit, 2007; Maury et al., 2016; Tsai et al., 2013). To our knowledge, an ideal animal model for listeriosis has not been established. Continuous new insights into animal physiology have increase the possibilities for infection systems, yet no single animal model completely aggregates the desirable characteristics to study human listeriosis. Therefore, the selection of an in vivo system is according to the specific objectives of the research being conducted.
Although animal models bring unquestionable insights into the study of infectious diseases, their extensive and indiscriminate use is strongly condemned by the European commission. This authority bases its policy on the Three R’s principle (Replacement, Reduction and Refinement), which aims to replace the use of animals with non-animal strategies, to use a reduced number of animals per experiment without compromising the ultimate aim of the research, and to improve practices that contribute to the welfare of animals from birth to death. When animal replacement is not possible, the use of animals must follow strict guidelines set out in EU Directive 2010/63/EU (European Parliament, 2010; Zuang et al., 2024).
4.1.1 Mammalian models (mice)The establishment of Robert Koch postulates to determine the etiological agent of an infectious disease, marked the inception of using mammalian species, phylogenetically related to humans, as healthy susceptible models (Kaito et al., 2020; Short and MacInnes, 2022).
Listeria monocytogenes is a ubiquitous microorganism, which enables it to infect a wide range of animals (Kammoun et al., 2022). However, in addition to humans, it mainly causes disease in ruminants, which, in an immediate and logical thought, should be the primary models to study listeriosis. However, this brings up many limitations. Thus, mice are the standard in vivo model to study listeriosis due to their size, ease of breeding and reproduction, rapid acclimation to confinement and an equivalent physiology when compared to humans (Lecuit, 2007). Commonly, mice are intravenously infected with the pathogen, and the role of some virulence factors, such as ActA and LLO, have emanated from this technique (Disson et al., 2009). Although mice are widely used in L. monocytogenes studies, the efficacy of oral infection is low due to the species-specific associated of with mammalian cells. As mentioned above, InlA binds to the E-cad receptor, which is a specific linkage for each species, depending on its 16th amino-acid type. Permissive species, such as guinea pigs, rabbits, humans and gerbils, have a proline in this position while non-permissive species have a glutamic acid – mice and rats have the glutamic acid and, consequently do not allow InlA binding (Lecuit et al., 1999). On the other hand, InlB naturally binds to C-Met in mice, humans and gerbils (Khelef et al., 2006). Theoretically, animals that naturally possess the imperative requirements to be bound to L. monocytogenes internalins, such as ruminants, non-human primates, and gerbils, should be selected to study listeriosis (Kammoun et al., 2022). Nonetheless, the ethical hurdles do not allow their wide application, so humanized mice have surged to overcome this limitation (Disson et al., 2008; Lecuit et al., 2001). Additionally, a “murinized” L. monocytogenes strain was developed to interact more closely with mouse E-cadherin. This modification involved altering the inlA gene in the L. monocytogenes EGDe strain to successfully infect wild-type mice (Wollert et al., 2007). Although this species-specific limitation was overcome, it was further discovered that the altered InlA was able to interact with both E-cadherin and N-cadherin in mice, luminally accessible in goblet and M-cells respectively, leading the bacteria to target both cells, increasing gut inflammation and consequently, hindering the capacity of L. monocytogenes to spread in the host (Tsai et al., 2013).
Currently there has not been a published comparative analysis of clonal complexes and their virulence in some animal models, such as gerbils, non-human primates, guinea pigs or rats. Although gerbils are permissive to both receptors, their use in listeriosis studies is limited. This may be related to the decreased sensitive to oral infection with L. monocytogenes when compared to other models, insufficient characterization when compared to mice and guinea pigs, absence of genetic models, and limited specific reagents and antibodies. The guinea pig infection model is advantageous in maternal-fetal studies as its placenta is the most comparable to human placenta among all rodents and has equivalent placental tropism. However, its narrow use may be related not only to species-specificity but also due to different disease symptoms from human listeriosis, with weak central nervous system tropism. Guinea pigs also present long gestation periods compared to mice, lack of gene deletion and transgenic models, and their larger size is more costly, limiting the number of animals per experiment. Despite the similarities of rats to mice, this infection model has shown low susceptibility to infection, requiring high infection doses to provoke disease. The use of non-human primates, as expected, is limited due to extended gestation periods, reduced number of available animals per study, and limited gene libraries compared to mice. Additionally, all these models are most costly when compared to mice (Cossart, 2011; D'Orazio, 2014; Eallonardo and Freitag, 2024; Hoelzer et al., 2012; Khelef et al., 2006; Roulo et al., 2014; Yan et al., 2023).
Considering this, very few articles have employed mice to investigate this phylogenetic association, either directly or indirectly (Domínguez et al., 2023; Maudet et al., 2022; Soni et al., 2017). Although the objective was not to compare strains from different CCs, Soni et al. (2017) inoculated three strains from CC1 in mice and observed a varying disease-causing capacity. One strain did not kill any mouse, while the other two presented 60 and 100% relative virulence. This highlights that although CCs are a more thorough classification, strains within a single CC can exhibit different virulence potentials. They also observed that the three strains harboured the major virulence genes, with the strain showing the lower pathogenicity presenting mutations in crucial virulence factors, such as listeriolysin O. However, no conclusion has been reached as to which mutation or genes better explain this unequal pathogenicity between phylogenetically close strains (Soni et al., 2017). Furthermore, in 2019, a large outbreak of listeriosis occurred in the Andalusian region, causing 207 cases, which was later associated with the strain ST388 from CC388 (Ministerio de Sanidad Consumo y Bienestar Social de España, 2019). Domínguez and her colleagues proceeded to investigate the virulence potential of this strain by comparing it with other strains from hypervirulent CCs (CC1 and CC4). In vivo infection assays were performed, and mice were infected intravenously with four strains (reference ATCC® 19115™, CC1, CC4, and CC388 strains). The results showed no significant differences between the CC388 strain and the other hypervirulent strains, as CC4 and CC388 isolates exhibited identical infection and spread ability (Domínguez et al., 2023). Maudet et al. (2022) selected strains from CC1, CC4, and CC6 – previously characterized as highly neuroinvasive CCs (Maury et al., 2016) – to perform infection assays in humanized KIE16P mice. The comparative analyses performed between these hypervirulent CCs and EGDe strain (CC9), corroborated the increased capacity of hypervirulent CCs to invade mice brains. Additionally, gene expression assays showed that hypervirulent strains presented upregulated levels of the inlAB operon, when compared to EGDe. Throughout these experiments different ∆inlB mutant strains were constructed to validate its relevance in the neuroinvasion capacity of L. monocytogenes. Despite the reduced neuroinvasion levels of EGDe when compared to CC4 strains, this study showed that whether using hypovirulent or hypervirulent strains, the inlB gene deletion reduced bacterial loads in the brain, confirming the need of overexpressing the inlB gene in L. monocytogenes neuroinvasiveness. Furthermore, the authors reported that InlB has immunosuppressive properties that are crucial to protect infected cells from host immune responses, resulting in an increase of infected monocytes’ lifespan and L. monocytogenes propagation to the brain. Additionally, as hypervirulent strains exhibit overexpression of inlB and are mainly associated with infections in immunocompetent individuals, this article highlights the need to continuously study hypervirulent CCs to improve our perspective regarding the bacterial factors employed in L. monocytogenes infection mechanism. Altogether, these finding showed that regardless of some reports suggesting strain-dependence in L. monocytogenes virulence studies, strains from hypervirulent CCs confer a significant concern to human health with distinct virulence factors that allows them to evade the host immune system. Moreover, mice models have proven to be a reliable tool to study L. monocytogenes infection cycle in mammals and we believe they will continue to be useful in future works.
4.1.2 Non-mammalian model organismsAlthough mammalian models are the paradigm for studying host-pathogen interactions, they still present many obstacles, such as ethical issues due to animal welfare, high costs, adequate facilities and differentiated training requirements. Therefore, alternative models are needed for in vivo experiments that are less costly, easier to manipulate, with a short life cycle and are ethically acceptable. The complexity and relevance of these models lie between the sophisticated humanized mice and the simplicity of in vitro approaches (Lecuit, 2007). A variety of invertebrate and vertebrate models have been used to study the virulence potential of pathogens and the host immune response (Ahlawat and Sharma, 2022; Mylonakis et al., 2007). In L. monocytogenes studies, we highlight the use of G. mellonella larvae, Drosophila melanogaster, Caenorhabditis elegans and Danio rerio (zebrafish), which have given valuable insights in the study of listeriosis. For instance, C. elegans model was previously used to evaluate the effects and toxicity of antimicrobial or antibiofilm substances in host-pathogen interactions and study nitrogen metabolism of L. monocytogenes after nematodes gut colonization (Kern et al., 2016; Muthulakshmi et al., 2022; Silva et al., 2015; Sivaranjani et al., 2016). The pathogenesis of L. monocytogenes has also been explored in D. melanogaster model, focusing in host immune system modulation, fly’s metabolism alterations upon infection, association between the bacterial growth dynamics and host’s genotypes (Chambers et al., 2012a,b; Hotson and Schneider, 2015; Mansfield et al., 2003; Taillebourg et al., 2014). Besides the widely use of both C. elegans and D. melanogaster models in the study of pathogenic bacteria, to our knowledge there are no comparative studies between L. monocytogenes CCs. In the fly model this can be related to the fact that the favourable temperature to flies is between 22 and 25°C, however, listeriosis studies are mainly conducted at 30–37°C and its inadequacy to distinguish between avirulent and virulent Listeria spp. after bacterial injection into the flies thorax (Jensen et al., 2007). On the other hand, C. elegans limited use may be associated to the fact that the deletion of some bacterial virulence genes (i.e., ActA) did not affect nematode’s death and that the C. elegans intestine architecture may be different from mammals, since neither cell junction during cell extrusion or in goblet cells lumen are common in this nematode (Balla and Troemel, 2013; Thomsen et al., 2006). Considering this, we focused on both zebrafish and waxworms to give an overview about the study of virulence potential between L. monocytogenes strains/CCs in non-mammals.
4.1.2.1 Insect modelsIn the past, it was thought that insects were not a good in vivo model to study microorganisms that cause disease in humans since they are not phylogenetically close. However, they share a few physiological aspects with humans. Human pathogens present an analogous virulence capacity in humans and insects, with similar virulence factors involved (Mansfield et al., 2003; Martinez et al., 2017; Mukherjee et al., 2010; Tsai et al., 2016). In addition, the pathogen follows similar infection cycle steps in both hosts. Consequently, insects have evolved some defence mechanisms that are shared between mammals and insect hosts, for instance, the innate immune system with physical and phagocytic barriers, that have a homologous function (Kemp and Massey, 2007; Peterson et al., 2008). However, insects lack the capacity to develop an adaptive immune response, which is a common feature in vertebrates (Ahlawat and Sharma, 2022; Tsai et al., 2016). Hence, insects as host models have been a convenient alternative to mammals for infectious disease research.
4.1.2.1.1 Galleria mellonella as an infection modelExperiments with Galleria mellonella larvae have been carried out for some time, with increasing interest in recent year as a potential surrogate model to explore pathogen infections (Dinh et al., 2021). Besides being small, cheap, short life cycle, easy to maintain and to obtain in large numbers, it is also adapted to temperatures from 25°C to 37°C – the optimum growth temperature for the vast majority of human pathogens (Dinh et al., 2021; Mylonakis et al., 2005). The wax worm is selected stage to be utilized as a model, with infection normally occurring by injection, which requires minimal training (Singkum et al., 2019; Tsai et al., 2016). Its whole genome has recently been sequenced, enabling the search for further novel insights (Lange et al., 2018). Moreover, these insects possess a relatively advanced innate immune system, comprising two main components – the cellular and humoral immune response. The primer is composed of hemocytes – phagocytic cells that prevail in the hemolymph, and they are also capable of encapsulation and nodulation of pathogens. The humoral response results from the production of lytic enzymes, antimicrobial peptides (AMPs), opsonins and melanin upon microbial exposure (Boman and Hultmark, 1987; Kavanagh and Reeves, 2004; Pereira et al., 2018). It has been reported that G. mellonella larvae infected with L. monocytogenes are prone to produce AMPs such as galiomycin, lysozyme, gallerimycin, insect metalloproteinase inhibitor (IMPI) and cecropin D (Mukherjee et al., 2011; Mukherjee et al., 2010). Beside the analysis of host’s immune modulation through hemocytes enumeration and variations in AMPs expression, the melanization, survival capacity, development of cocoon, motion ability can be evaluated in infected larvae with L. monocytogenes (Kavanagh and Sheehan, 2018).
Galleria mellonella has been utilized as an infection model to study the virulence potential of L. monocytogenes through comparative studies with different Listeria species or comparisons between L. monocytogenes serotypes (Martinez et al., 2017; Mukherjee et al., 2010; Pan et al., 2024; Rakic Martinez et al., 2020). Mukherjee and co-workers explored the ability of this insect model to discriminate between non-pathogenic and pathogenic Listeria species. When injected with 106 CFU/larva, strains belonging to non-pathogenic species, such as L. innocua and L. seeligeri were observed to have a lower infection capacity than the L. monocytogenes EGD-e strain; and, although L. ivanovii caused a significant but slightly higher mortality than the non-pathogenic species, it presented a reduced pathogenicity efficiency compared to L. monocytogenes (Mukherjee et al., 2010). These results were corroborated by Martinez et al. (2017), who observed that, at the same inoculum level, the L. monocytogenes LS1209 reference strain displayed a LT50 (lethal time to kill 50% of larvae) 4 to 6 times lower than the non-pathogenic Listeria strains (Martinez et al., 2017).
The wax model was used to test the virulence potential of L. monocytogenes strains of different serotypes. The serotype 4b strain, commonly associated with clinical cases, expressed the highest larvae killing rate and was more pathogenic than the serotype 1/2a strain, usually related to food isolates. Other serotypes tested, 4a, 4c and 4d, also showed a lower pathogenic potential (Mukherjee et al., 2010). However, the study conducted by Martinez et al. (2017) showed that strains from different serotypes (1/2a, 4b, 1/2b) resulted in similar larvae mortality and identical LT50 at 24 h when administered at 106 CFU/larva (Martinez et al., 2017). This lack of correlation between serotypes and virulence potential was in clear contrast to the findings of the former study, highlighting the importance of considering potential confounding. These may include differences in the dose of bacteria injected (106 CFU/larva in the first study, whereas three different concentrations – 106 CFU/larva, 105 CFU/larva and 104 CFU/larva – were used in the second) and the parameters analysed (Mukherjee et al. monitored the % survival along 7 days, whereas Martinez et al. focused on LT50 at 24 h and % mortality – not specifying its progression over the infection period). Nonetheless, it was concluded in both studies that the virulence potential of L. monocytogenes is dose and strain dependent, so these different results could be explained by the use of different L. monocytogenes strains. Another factor that could externally influence on the observed results is the larvae’s diet, since no information was available on the rearing of the larvae used in the Martinez et al. research. Previous studies have shown the importance of the diet in the larvae development, health, hemolymph volume and hemocyte concentration, which subsequently affect the immune response of G. mellonella (Jorjão et al., 2018; Kwadha et al., 2017). It has also been published that the diet of worms has an important impact in microbiological studies (Banville et al., 2012; Jorjão et al., 2018). Hence, standardization of diets could reduce external biases on results allowing for interlaboratory comparisons.
To date, virulence evaluation of different L. monocytogenes CCs using G. mellonella has only been performed by Cardenas-Alvarez et al. (2019). This insect model was used to compare the pathogenic potential of CC1, CC6, CC7, CC9, CC14, CC37, and CC204 strains. Briefly, differences were observed between strains from different CCs, with strains from the putatively hypervirulent CCs, CC1, and CC14, causing a reduced average survival rate (33.2 and 29.1%, respectively). Oppositely, isolates from CC9, widely accepted as hypovirulent CC, presented the highest survival rate (53.5%). In addition, the remaining CCs (6, 7, 37 and 204) showed an intermediate range of survival rates from 40 to 50%. Another parameter evaluated was the LD50 value (median lethal dose) – calculated from the colonies counted on plates and the number of larvae killed per day – lower values were observed for CC14, meaning that fewer cells of the pathogen are needed to kill G. mellonella. Cytotoxicity was also evaluated by measuring the level of lactate dehydrogenase (LDH), which is a signal of cell damage after bacterial infection. CC14 strains caused significantly less cytotoxicity than other CCs (CC6 and CC7). A positive correlation was found between LD50 and cytotoxicity, therefore CC14, strains by having a reduced LD50, also caused less injuries to host cells, which is hypothesized to be a defence mechanism to escape the host immune system and successfully spread (Cardenas-Alvarez et al., 2019). Considering these results, G. mellonella as an infection model, besides the capacity to differentiate non-pathogenic from the pathogenic Listeria species, has the potential to distinguish between virulent and attenuated L. monocytogenes strains from different CCs, validating its ability to discriminate the virulence potential of L. monocytogenes.
4.1.2.2 Zebrafish modelThe non-mammalian vertebrate Danio rerio, known as the zebrafish, is an in vivo model that has been gradually catching the attention of researchers for the study of infectious diseases as it meets the ideal features of vertebrate and mammalian models (Shan et al., 2015). As other models already described in this review, zebrafish is more easily applicable and economically and ethically acceptable than most mammalian models. Being a vertebrate, its morphological and genetic similarities with humans are more pronounced than with invertebrate (Pont and Blanc-Potard, 2021). In addition to the zebrafish’s large clutch dimension, ex-utero growth and small size, its transparency makes zebrafish a distinctive mode, allowing observation of the early stages of growth and enabling the real-time observation of bacterial infections (Pont and Blanc-Potard, 2021; Shan et al., 2015; van der Sar et al., 2004). Another interesting peculiarity is that the innate and the adaptive immune systems are temporally separated, where the primer acts singularly during early weeks while the latter is perceived just during the 4–6 weeks post-fertilization (Herbomel et al., 1999; Herbomel et al., 2001; Lam et al., 2004; Trede et al., 2004).
The use of this vertebrate model in the study of host-pathogen interactions began in 1999, when Philippe Herbomel et al., reported that primitive macrophages – which evolve during the embryo’s development and subsequently give rise to hematopoietic stem cells – develop in the zebrafish embryos at 22 h post-fertilization (Herbomel et al., 1999). Therefore, zebrafish have been used to explore host-pathogen interactions and provide new insights into the capacity of L. monocytogenes to cause disease in this in vivo model (Levraud et al., 2009; Shan et al., 2015; Zakrzewski et al., 2020). The different stages of development of zebrafish are used for research and have their advantages, but infection assays have only been performed in zebrafish’s embryos to study the association of clonal complexes with hyper-and hypovirulence of L. monocytogenes strains, thus, our review will merely focus on this developmental stage. Among these, Hurley et al. (2019) made use of L. monocytogenes strains collected from three meat and vegetable processing facilities over 4 years. Genome analysis on these isolates reported distinct virulence genotypes and grouped them into hypervirulent, hypovirulent and unknown virulence groups (Hurley et al., 2019). This classification was slightly different from that previously described by Maury et al. (2016), as the isolates commonly associated with clinical cases (strains from CC1, CC2 and CC6) were underrepresented among the isolates collected. Therefore, hypervirulent strains were selected based on the presence of additional virulence factors such as listeriolysin S from LIPI-3 or LIPI-4. Selected hypovirulent strains (CC121, CC9, CC31) harboured PMSC mutation in the inlA gene and some of them had a deletion on the actA gene, which is associated with a decrease in intracellular spread. Isolates with integral virulence factors or with minimal mutations in some genes were classified as having unknown virulence capacity (CC3). Zebrafish embryos infected with putatively hypervirulent strains presented only a 3% survival rate, followed by zebrafish embryos infected with isolates of unknown virulence (20% survival rate), while hypovirulent strains caused a higher survival rate of 53–83%, requiring 72 h post-infection to cause this decline. Using the zebrafish infection model, Hurley et al. (2019) were able to discriminate the different virulence phenotypes and confirm the previous virulence genotypes obtained by WGS (Hurley et al., 2019). Muchaamba et al. (2022) also performed infection assays using the zebrafish embryo model, comparing the virulence potential of L. monocytogenes strains by lineage, serotype, and clonal complex. When the strains were grouped by CC, the researchers observed virulence discrepancies by CC and strain-specific intra-clonal complex. Embryos infected with CCs that are generally considered hypervirulent showed higher mortality than isolates from CC9 or CC8. Within some CCs, such as CC1 and CC9, strain-dependent virulence variation was observed – three CC1 strains required more than 24 h post-infection to cause 100% mortality, and while two CC9 strains exhibited no virulence, the other three CC9 strains presented variable levels of virulence. The conclusion of the in vivo assays was that the virulence potential of this pathogen varies with genotype, serotype and strain (Muchaamba et al., 2022). Therefore, both studies confirmed the previous categorization of hypervirulent and hypovirulent L. monocytogenes CCs using the zebrafish embryo infection model. This underscores the model’s relevance as an in vivo tool for further elucidating the virulence phenotypes of L. monocytogenes strains.
4.2 In vitro infection modelsThe in vitro systems represent alternative processes to study bacterial virulence as they mimic the infectious mechanism, allowing, for example, screening of pathogen gene expression and how the deletion of some genes affects the behaviour of strains in physiological environments mimicking in vivo conditions. In vitro assays are based on the assumption that pathogens, such as L. monocytogenes, have the ability to infect hosts by attachment, invasion, multiplication and subsequent dissemination in either phagocytic or non-phagocytic cells through the production of virulence factors (Liu et al., 2007). Although these systems do not precisely replicate the full features of the host-pathogen interaction, as infectious agents may encounter unfavourable conditions and the host immune system, when compared to in vivo models they are less expensive, less time consuming and less ethically demanding, allowing large-scale experiments. Additionally, the ability to control experimental conditions allows to unravel favouring factors in disease. Therefore, their use is recommended for preliminary studies to find new virulence factors, after which in vivo models can be used on a limited scale to confirm the results (Lehr, 2002; McCoy et al., 2024; Chiang et al., 1999). For these reasons many different in vitro models have been developed. The standard in vitro system, that has been used for decades, is the 2D monolayer culture of immortalized human cells. More recently, in a bioengineering context, there has been an increase in the use of different systems based on in vitro and ex vivo models, such as organoids and 3D cell cultures, to improve the monolayer model (Taebnia et al., 2023). The choice of an appropriate in vitro model should focus on the definite biological issue, for example, cell lines are more adequate to study precise interaction processes of pathogens. The addition of unneeded complexity can be disadvantageous, shrouding relevant host-pathogen interactions (McCoy et al., 2024). Therefore, many tissue culture experiments to study adhesion, invasion, cell to cell spread in different cell lines, survival in macrophages, evaluation of cytotoxicity and pathogens activity upon different host environmental conditions (e.g., pH and temperature) have been reported to describe and determine novel virulence concepts of bacteria (Conte et al., 1994; Hasebe et al., 2017; Wagner et al., 2022). In L. monocytogenes, in vitro models have been used to investigate host-pathogen interactions at either an intestinal, cerebral or placental level. These models brought significant knowledge regarding the L. monocytogenes intracellular cycle, invasion at cell extrusion sites, the role of putative virulent genes in cell invasion, required internalins (InlA, InlB and InlP) to placental invasion, L. monocytogenes bacteriocins in intestinal commensals, pathogen’s routes to invade the brain and other related aspects (Banović et al., 2020; Cabanes et al., 2004; Cabanes et al., 2005; Lamond and Freitag, 2018; Pentecost et al., 2006; Rolhion et al., 2019).
4.2.1 Tissue culture assays for adhesion, invasion, intracellular growth and cell-to-cell spreadIn 1948 the first cell line based on subcutaneous mouse tissues was developed. Thenceforth, various mammalian cell lines have been developed and used as the primary in vitro model to investigate infectious diseases, since they mimic host defence mechanisms (Magdalena, 2017). In L. monocytogenes, the human colorectal adenocarcinoma cell line Caco-2 is one of the most popular cell models that replicate the intestinal barrier, along with HT-29, Henle-407, HeLa, and many other cell lines (Liu et al., 2007; Pizarro-Cerdá et al., 2012). Different cell lines used in listeriosis studies were represented in Table 3.
留言 (0)