
記住我
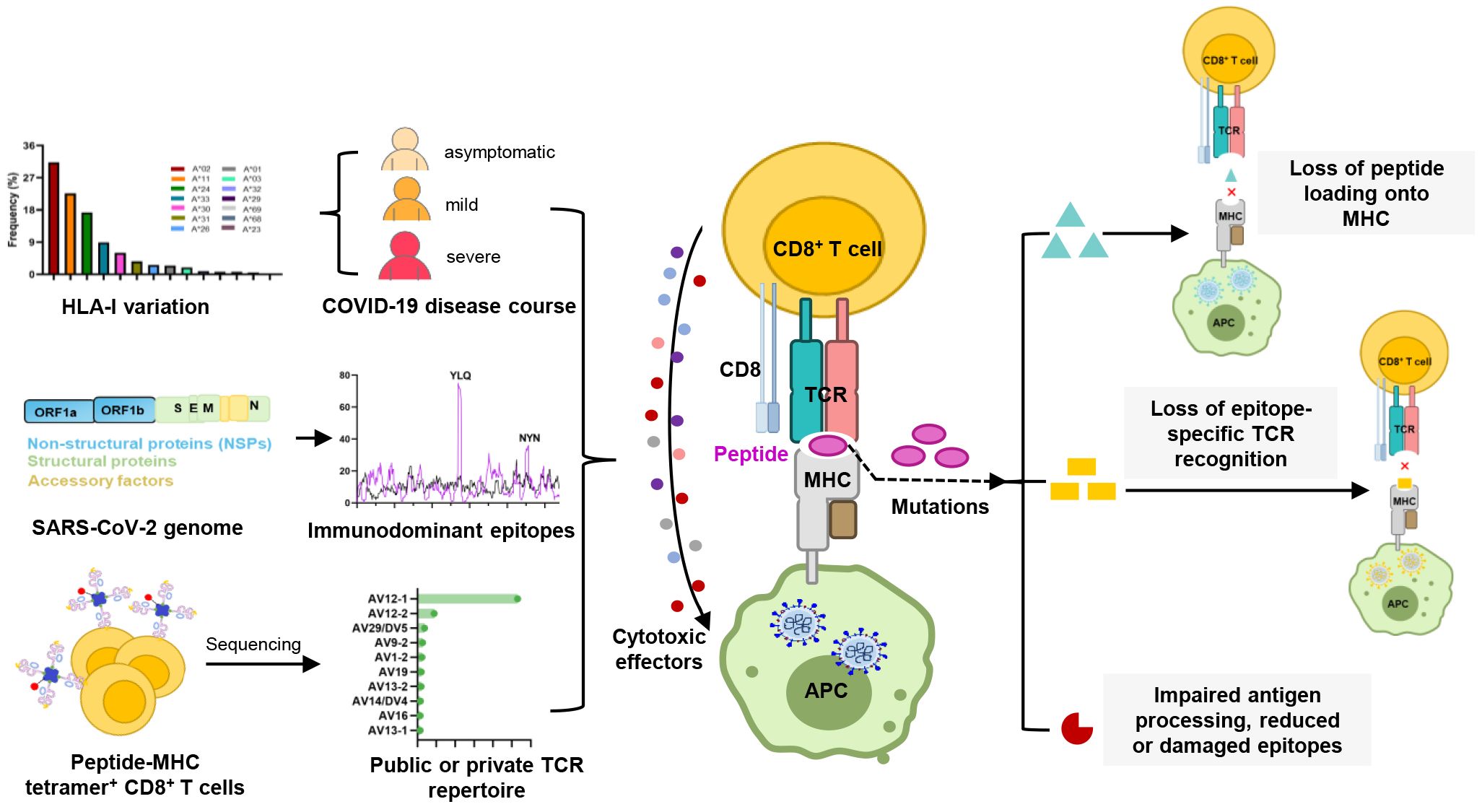
Graphical Abstract.
1 IntroductionIn late 2019, the emergence of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) as a novel human coronavirus drew global attention to the fight against this infection (1). SARS-CoV-2 infection causes COVID-19 disease, with clinical presentations ranging from mild or asymptomatic to severe and fatal respiratory illness (2). Approximately 15% of confirmed cases are classified as severe, most occurring in individuals over 65 years of age or those with underlying medical conditions (3).
During SARS-CoV-2 infection, neutralizing antibodies, CD4+ helper T cells, and CD8+ killer T cells all contribute to controlling the virus and providing protection against viral pathogens (4, 5). Unlike the transient and heterogeneous nature of neutralizing antibodies (6–8), T cells play a critical role in conferring immune memory and establishing long-term memory responses (9–13). For closely related coronaviruses, such as SARS-CoV-1, memory T cell responses have been detected up to 17 years after infection (14, 15). Most COVID-19 convalescent patients exhibit broad and robust SARS-CoV-2-specific T cell responses (11–13, 16–20). It has been reported that functional SARS-CoV-2-specific T cell responses are retained at 6 months following infection (21, 22). Moreover, SARS-CoV-2-specific T cell responses were detectable in antibody-negative patients or in patients with B cell-deficient agammaglobulinemia, suggesting that T cells may effectively respond to the virus even in the absence or insufficiency of antibody responses (11, 16). Consistent with the positive effects observed in convalescents, the reduction of T cell lymphocytes is often used as a marker of disease severity (17, 18). Although circulating SARS-CoV-2-specific CTLs are less consistently observed than CD4+ T cells (4, 10, 13), their presence is generally associated with better COVID-19 outcomes (4, 12). The proportion of multifunctional CTLs is higher in mild patients compared to those with severe symptoms, further highlighting the positive role of CTLs in mitigating disease severity (12). Both in vivo and in vitro studies have demonstrated significant activation of CTLs during SARS-CoV-2 infection (10, 17, 23, 24).
The breadth and nature of the cellular immune response to viral infection are driven by the diversity of the T cell receptor (TCR) and major histocompatibility complex (MHC, HLA in humans). The tripartite interaction of TCR-peptide-MHC (TCR-pMHC) forms the basis for CTL responses against viral infections and malignancies, while maintaining auto-tolerance and averting autoimmune diseases. The antigen specificity of CTL responses is influenced by the expression of host HLA class I (HLA-I) alleles, each of which presents a virus-derived peptide ranging from 8 to 11 amino acids in length (25). Certain locations within antigenic epitopes, also described as anchor residues, have been shown to be critical for antigen presentation, and mutations in these anchor residues may disrupt peptide binding to HLA-I molecules (26, 27). Moreover, mutations within epitopes may also impact the interaction of TCRs with antigenic peptides, as seen with the P272L mutation in the YLQ (A*02/S269-277, YLQPRTFLL) epitope and the Y453F mutation in the NYN (A*24/S448-456, NYNYLYRLF) epitope (28, 29). Such mutations may interfere with the TCR-pHLA tripartite interaction, potentially leading to the failure of T cell activation, a phenomenon known as T cell immune escape (20, 29–31).
Given the importance of TCR-pMHC interaction during SARS-CoV-2 infection, there is a particular need for in-depth studies and a comprehensive understanding of TCR-pMHC complex. However, the extent to which MHC polymorphism and TCR diversity, especially concerning epitope specificity, contribute to CTL responses remains ambiguous. In this review, we evaluated the published studies about CTL immune response against SARS-CoV-2, focusing on the epitope-presenting mechanism of TCR-pHLA complexes, HLA variation in the context of various COVID-19 disease outcomes, and the characteristic of peptide-specific TCR repertoire. We also discussed how SARS-CoV-2 variants of concern evade CTL immunity and emphasized potential strategies in response to Omicron and future variants, providing a basis for vaccine optimization and prevention of reinfection.
2 HLA-I variation and its association with COVID-19 disease courseHLA-I genes primarily encompass the classical, highly polymorphic HLA-A, HLA-B, and HLA-C genes, as well as non-classical HLA-E, HLA-F and HLA-G with limited polymorphisms. HLA variation directly affects the binding affinities of HLA molecules to antigenic peptides, thereby influencing the recognition of pathogen-derived antigens by immune cells (32–36). To date, pathogen-driven HLA selection has been proposed and demonstrated in studies of various infectious diseases. A well-documented example of HLA alleles influence viral infections is HIV (human immunodeficiency virus) infection. Certain HLA molecules, like HLA-B*27, B*57, and B*58:01, can accommodate specific HIV antigens and trigger effective immune responses (37). The progression of HIV infection is strongly associated with the homozygosity of HLA-I genes and differential HLA expression levels (38, 39). Additionally, HLA variation has been linked to hepatitis B, hepatitis C, and several other infectious diseases (40).
Given the pandemic nature of SARS-CoV-2 and the inherent difficulties in assessing the risk of infection, the most robust genetic association studies of SARS-CoV-2 infection have mainly focused on disease outcomes. Thus far, numerous studies have explored the association between HLA alleles and COVID-19 outcomes, but without a clear consensus. In fact, some large studies, either genome-wide association studies (41) or large HLA databases (42), have failed to show significant effects of HLA alleles on disease. Different populations may possess different alleles associated with susceptibility, depending on the HLA allele pool present in each population. In addition to study design and statistical differences, this may be a reason why no conclusive association between HLA and COVID-19 has been reported to date (43–52).
Nevertheless, some correlated findings have emerged and are summarized in Table 1. For instance, at least two studies have reported an association between HLA-A*01:01 and diminished CTL responses (53, 54). In two other independent studies, A*11:01 and B*51:01 were identified to be associated with severe COVID-19 disease (51, 55). HLA-C*04:01 has also been found to be associated with a severe clinical course of COVID-19, with patients carrying this allele having twice the risk of requiring mechanical ventilation (56). HLA-C*14:02 allele was significantly predisposed to the worst outcomes in COVID-19 patients (51). HLA-E*01:01 allele and heterozygous HLA-E*01:01/03 genotype are associated with severe COVID-19, which may account for the individual difference in NK cell responses following SARS-CoV-2 infection (57). HLA-B*15:27, B*27:07, and C*07:29 genotypes have been found at high frequencies in infected patients (46, 48). Schindler et al. found an association between the A*30:02 allele and viral infection (58). A comprehensive computer analysis of peptide-HLA-I binding affinities across over a hundred HLA-A/B/C genotypes revealed that B*46:01 has the fewest predicted binding peptides for SARS-CoV-2 (59). This suggests that individuals with B*46:01 may be particularly susceptible to COVID-19 (59), a conclusion also supported by an earlier study related to SARS-CoV-1 (63). Moreover, some HLA genotypes have shown strong associations with mild disease and cross-reactive CTL responses. A strong association was reported between B*15:01 and asymptomatic infection in patients capable of clearing the virus during the early stages of infection (60, 61). Predicted SARS-CoV-2 peptides presented by B*15:03 are highly conserved across all other human coronaviruses (HCoVs), implying the potential for cross-protective T cell immunity (59). HLA-A*02:05, B*58:01, and C*07:01 have been reported to be associated with protective effect against SARS-CoV-2 infection. Overall, despite mixed results, some consistent patterns are beginning to emerge in the association between HLA alleles and SARS-CoV-2 infection. These patterns may offer a valuable foundation for interpreting studies related to viral antigen presentation.
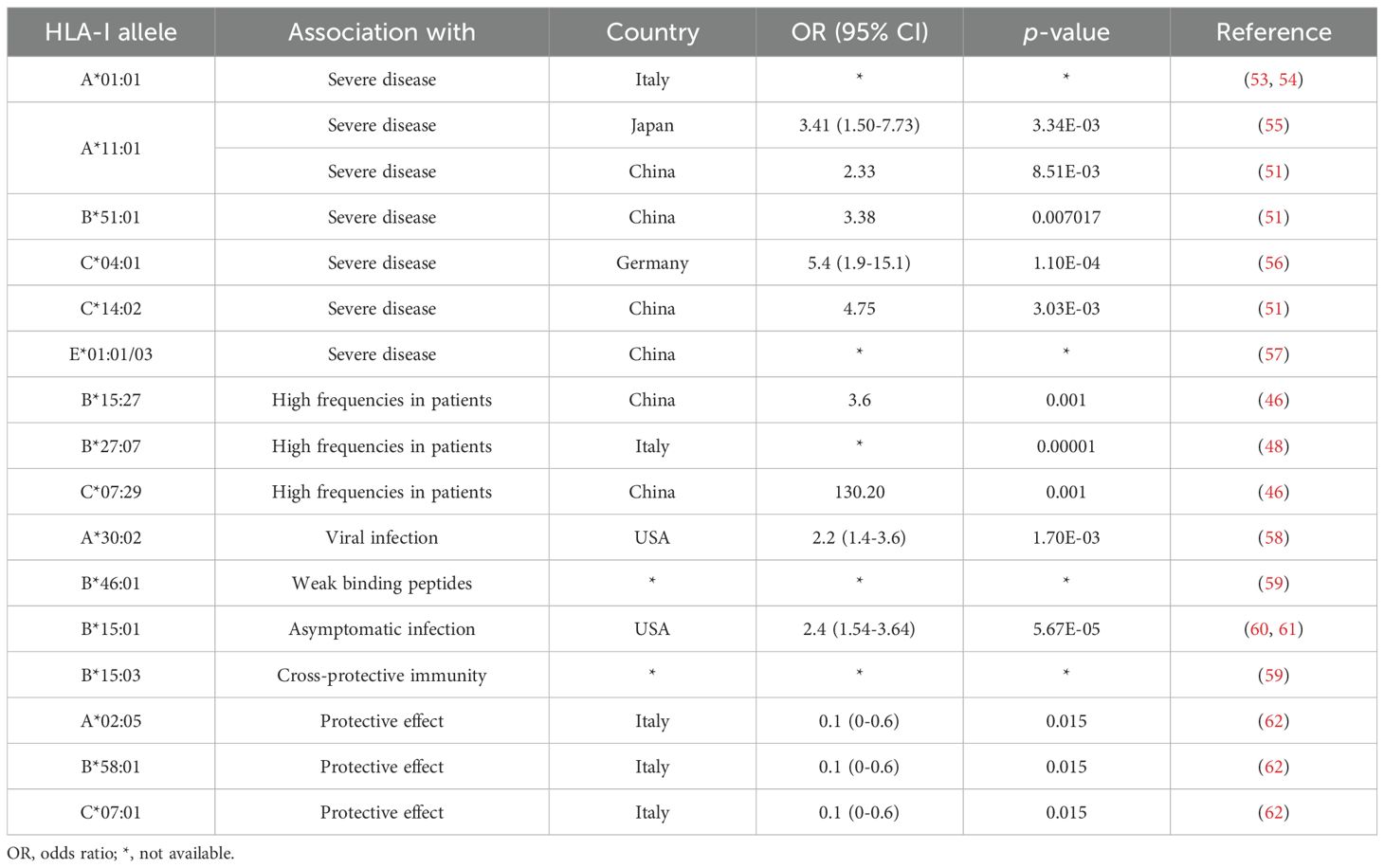
Table 1. Summary of HLA associations with COVID-19 outcomes.
3 Analysis of SARS-CoV-2 CTL epitope distribution and immunodominanceSARS-CoV-2 epitopes have been identified for over 30 HLA-I alleles, such as HLA-A*02:01, A*24:02, A*01:01, and B*07:02 (64). According to the information provided by Immune Epitope Database (IEDB, http://tools.iedb.org/immunomebrowser/) (65), CTL responses are directed at multi-antigen, encompassing structural proteins such as S (spike), N (nucleoprotein), and M (membrane protein), as well as non-structural proteins like ORF3a, ORF7a and ORF8. Viral proteins that are abundantly expressed in SARS-CoV-2-infected cells tend to be the most dominant targets in the T cell response to viral invasion (10, 11, 13, 66, 67). This phenomenon may be attributed to the fact that the immunodominant pattern of T cells against SARS-CoV-2 is closely associated with the expression level of viral proteins (10). To date, numerous studies have reported immunodominant T cell epitope. However, there are significant variations among these studies, including differences in screening procedures, HLA alleles considered, antigens targeted, sample sizes of individuals analyzed, and the criteria used to define “immunodominance” (12, 68–70). For example, Peng et al. reported several immunodominant peptides, defining them as those recognized by at least 6 individuals out of a pool of up to 16 subjects screened (12). Tarke et al. also highlighted the presence of highly immunodominant epitopes, with 49 HLA-II-restricted epitopes recognized by at least 3 out of an average of 10 donors, and 41 HLA-I-restricted epitopes recognized by over 50% of HLA-matched donors (68). Nielsen et al. found a broad spectrum of T cell responses, with the top three immunogenic epitopes derived from different SARS CoV-2 proteins (70). Keller et al. defined immunodominant epitopes as those recognized by multiple donors from M, N and S viral proteins (69).
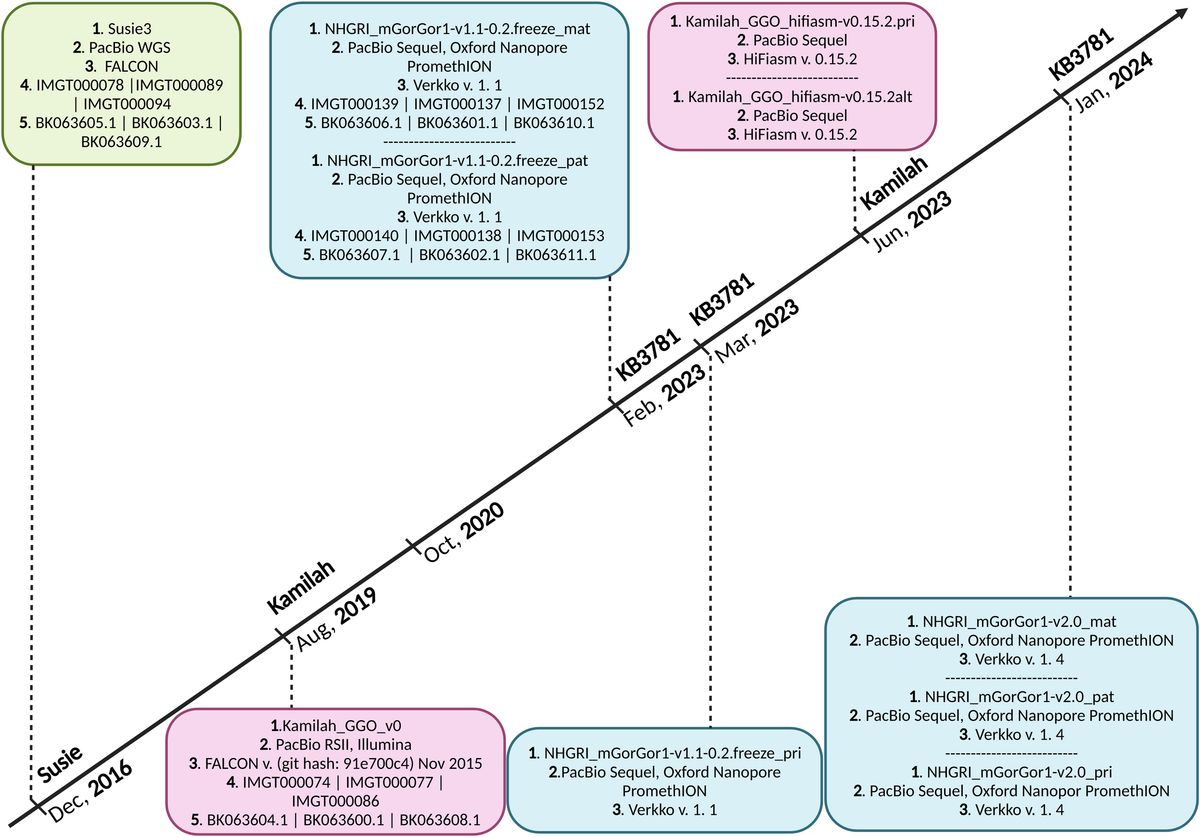
Several independent studies, including one involving the BNT162b2 mRNA vaccine, have identified the YLQ-epitope as immunodominant (20, 23, 71–74), eliciting an immune response in the vast majority of convalescents with the HLA-A*02 genotype (responses in 16 out of 17 individuals studied) (66, 70, 71). Another notable example is the HLA-A*01:01-restricted TTD epitope (A*01/NSP3819-828, TTDPSFLGRY). Nelde et al. reported a positive response to the TTD epitope in 83% of donors (67). Saini et al. conducted an extensive analysis of over 3,000 peptides for 10 HLA alleles and confirmed the recognition of the same HLA-A*01:01-restricted epitope (75, 76). Additionally, dominant CD8+ T cell responses have been identified for the LTD (A*01/S865-873, LTDEMIAQY) epitope and KCY (A*03/S378-386, KCYGVSPTK) epitope when analyzing vaccine-elicited CD8+ T cell responses that span the whole S protein (77). Using the Immunome Browser tool (developed and hosted by the IEDB, https://www.iedb.org/), we plotted the epitope assay counts for each residue of SARS-CoV-2 ORF1ab, S, N, M, and ORF3a proteins. The number of positive and negative assays are indicated for each residue position, allowing for the visualization of immunodominance patterns (Figure 1). A range of potentially immunodominant CTL epitopes were identified on these viral proteins, some of which have been characterized by peptide-specific TCR repertoires, including ORF1ab-TTD (67, 75, 76), spike-YLQ (20, 71, 78–82), NYN (79, 83, 84), LTD (79, 84), RLQ (71, 84) (A*02/S269-277, RLQSLQTYV), QYI (78, 79, 82–84) (A*24/S1208-1216, QYIKWPWYI), nucleoprotein-SPR (80, 82, 84, 85) (B*07/N105-113, SPRWYFYYL), MEV (82) (B*40/N322-331, MEVTPSGTWL), KTF (82, 86) (A*03/A*11/N361-369, KTFPPTEPK), and ORF3a-FTS (79, 84) (A*01/ORF3a207-215, FTSDYYQLY) (Table 2). Meanwhile, we also observed that some potentially dominant epitopes, such as membrane protein-GLM (A*02/M89-97, GLMWLSYFI) and ORF3a-FTS, have emerged several mutations associated with CTL immune escape, including GLM-L90F and FTS-Q213K mutations (20, 88) (Table 3). These dominant epitopes warrant further investigation, as they may offer valuable insights for developing effective SARS-CoV-2 vaccines.
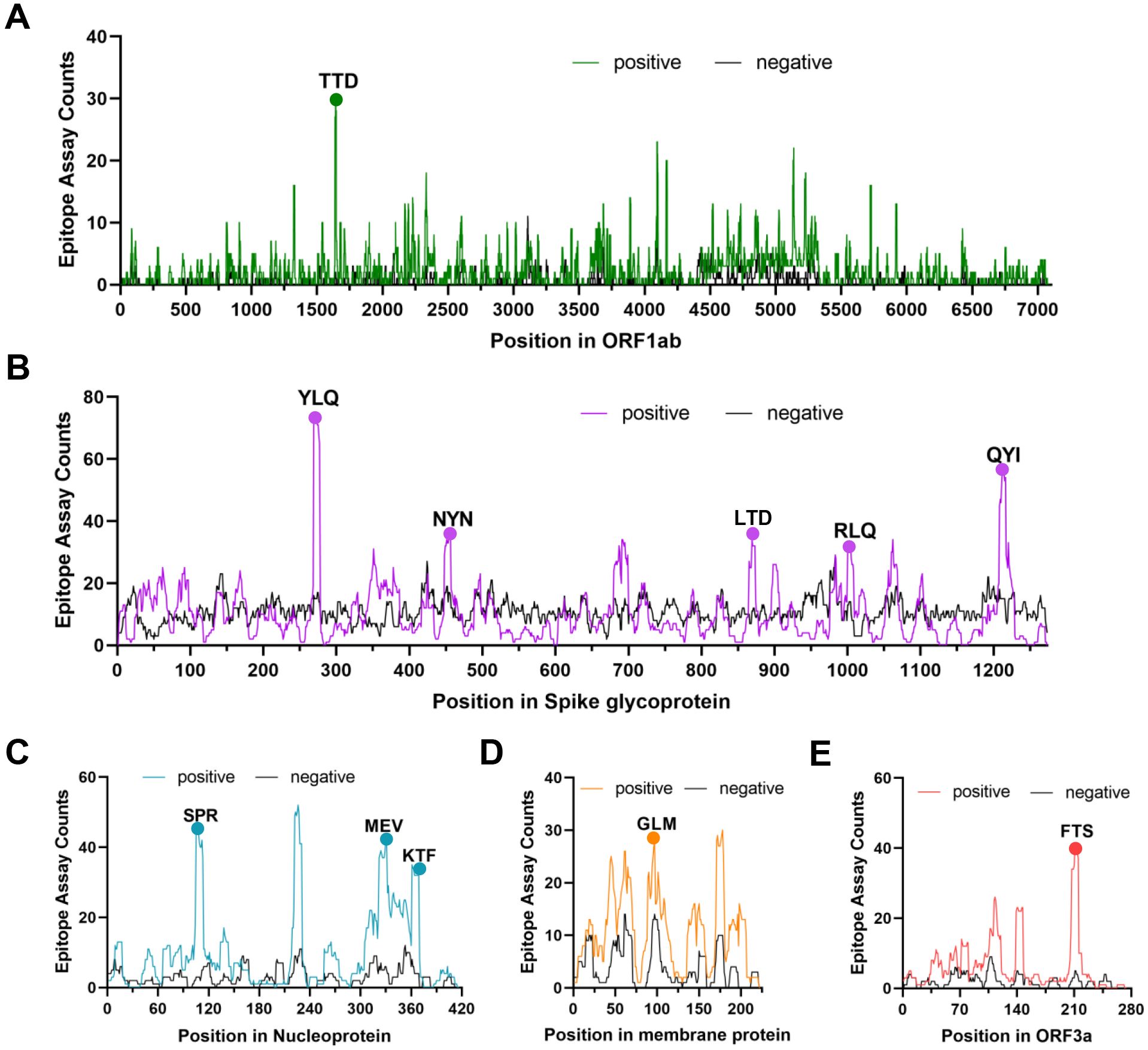
Figure 1. The identification of immunodominant antigenic regions of SARS-CoV-2 CTL epitopes. (A-E) CTL Epitope assay counts of five viral proteins (ORF1ab, Spike, N, M, and ORF3a) were analyzed using the IEDB’s Immunome Browser tool to identify potential antigenic regions. The number of positive and negative assays are indicated for each residue position.
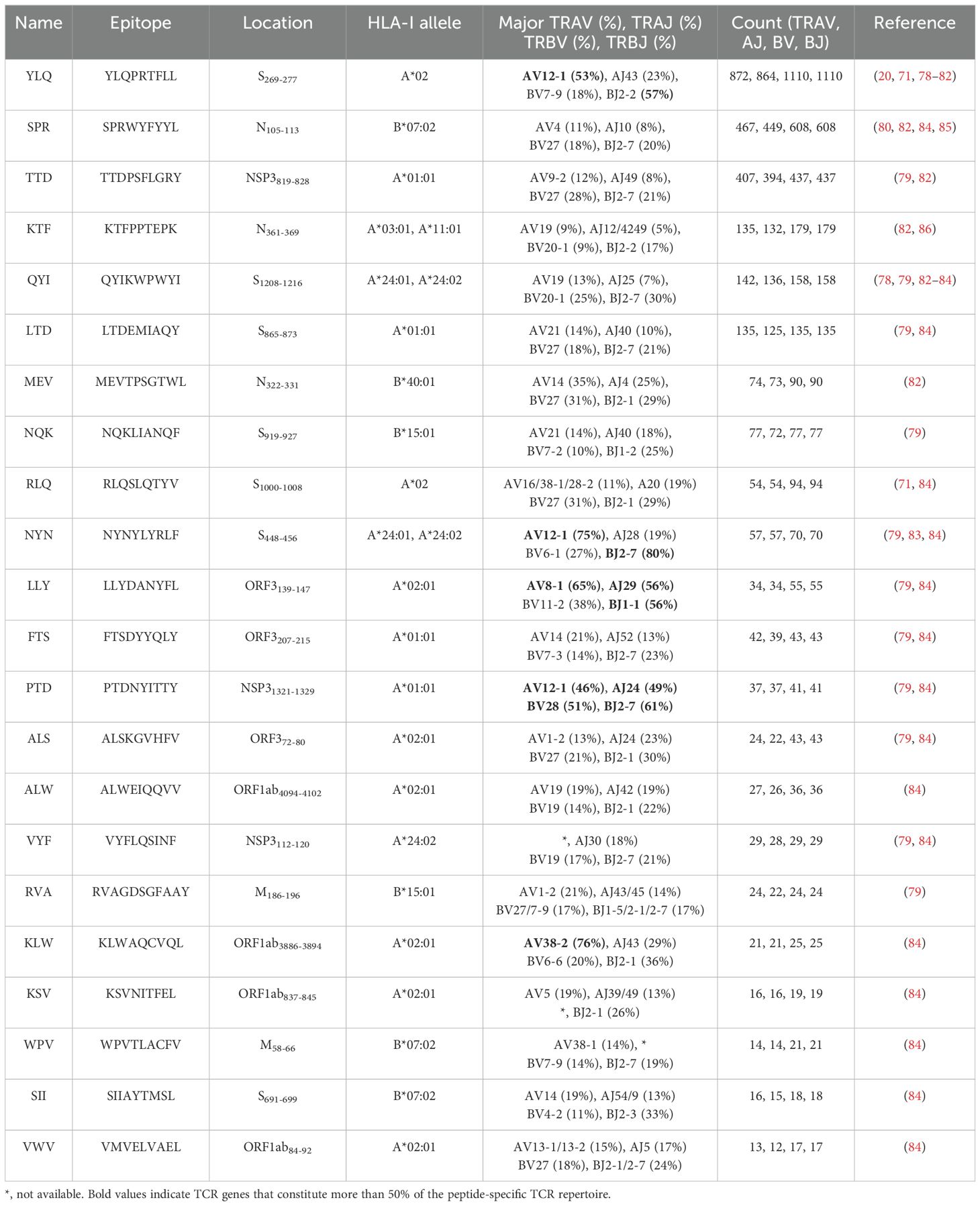
Table 2. TCR epitope characteristics of 22 SARS-CoV-2 epitopes in the public VDJ database (87).
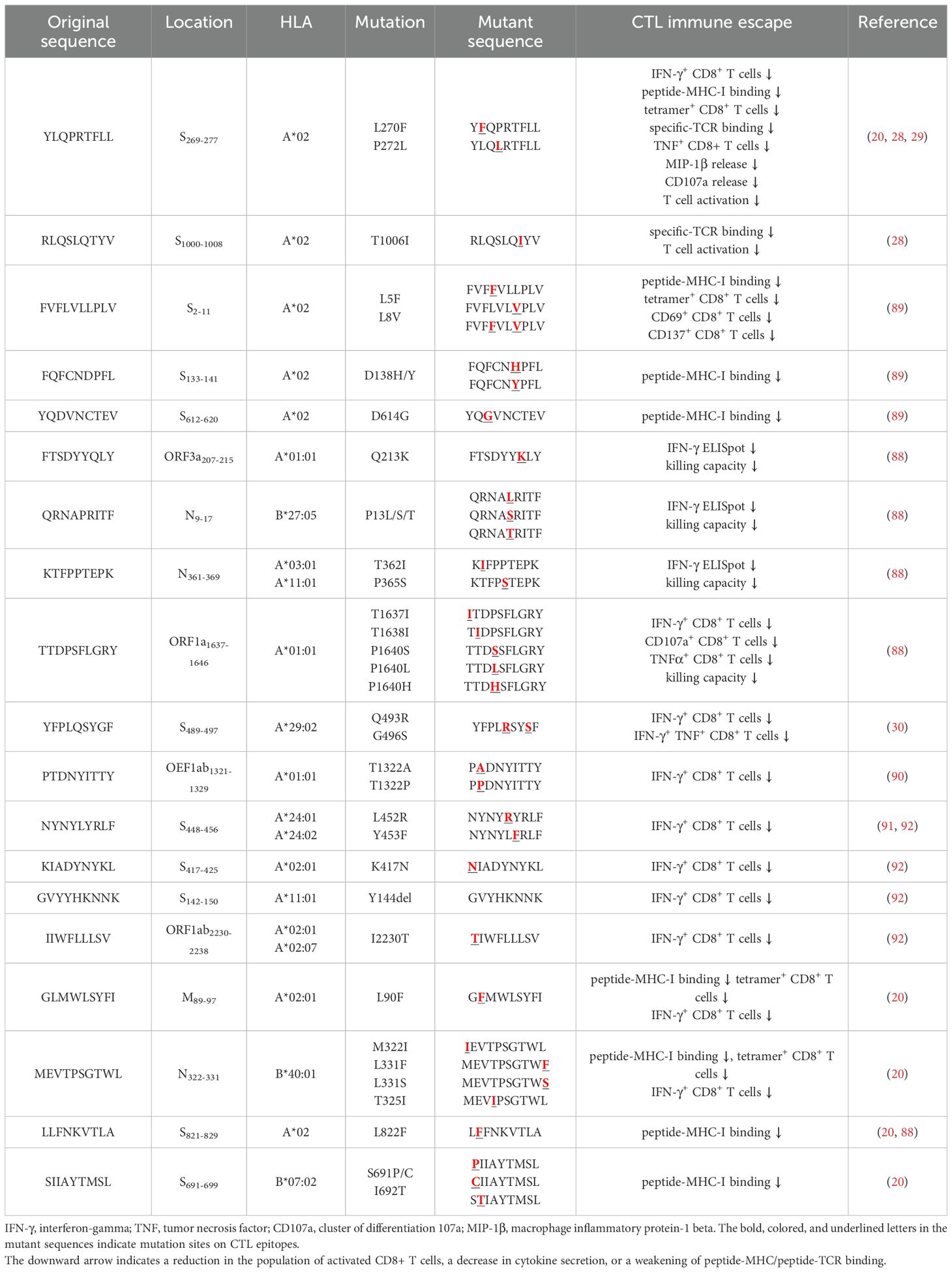
Table 3. CTL immune escape by SARS-CoV-2 mutations.
4 Biological insights into COVID-19 from the TCR repertoireThe specificity of T cells toward viral antigens presented by MHCs is determined by unique TCRs (93). TCRs exhibit considerable sequence heterogeneity due to somatic recombination of different variable (V), diversity (D), and joining (J) gene fragments, as well as random mutations of nucleotides at segment junctions (94). As a result, each TCR chain possesses three variable complementary determination regions (CDRs): the germline-encoded CDR1 and CDR2 loops, and the hypervariable CDR3 loop (Figure 2A). Upon antigen recognition, activated T cells undergo rapid clonal expansion, resulting in a substantial increase in T cells with identical TCRs, thus producing the identical antigen recognition (95). Expanded CTL clones possess specific TCRs for viral antigens, directly lyse infected cells via perforin/granzyme release, and secrete pro-inflammatory mediators (87). Hence, investigating the role of TCR in SARS-CoV-2 infection should garner vast interest.
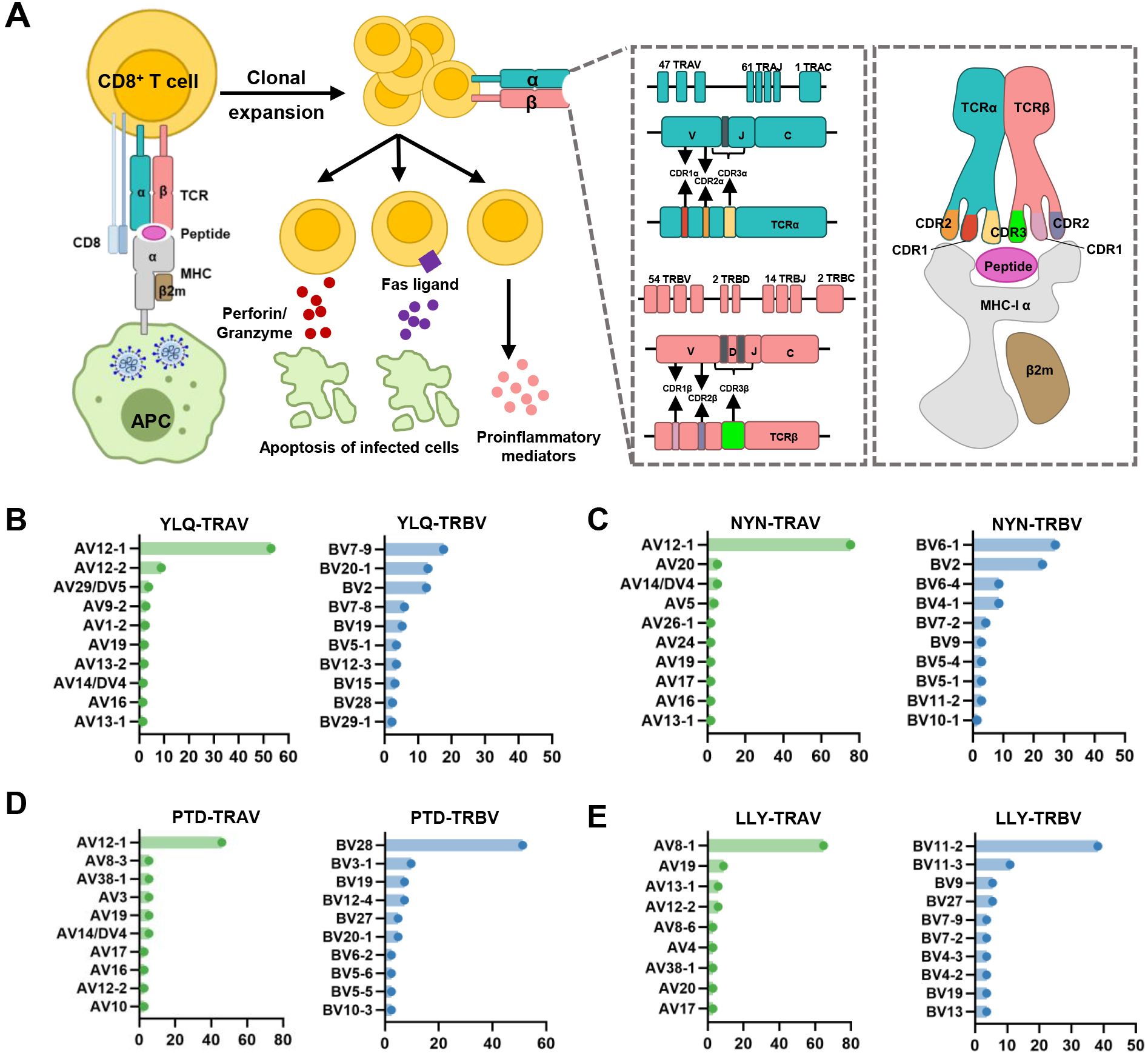
Figure 2. Overview of CTL response to SARS-CoV-2 infection mediated by TCR-pMHC complex. (A) Antigen presenting cells (APCs) endocytose SARS-CoV-2 and degrade it through antigen processing. These epitope fragments are then presented on the cell surface by MHC molecules and allow recognition by T cells. TCR genes of the α-chain (TCRα) and β-chain (TCRβ) on the T cell surface are recombined to produce a diverse TCR repertoire. If a CD8+ T cell is able to bind pMHC, it will undergo clonal expansion and directly target infected cells through perforin/granase, FAS ligand/tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) pathway, or secretion of pro-inflammatory mediators. CDR1 and CDR2 are encoded by TRBAV and TRABV genes, and CDR3 encompasses VJ regions (for TCRα) or VDJ regions (for TCRβ). Non-template nucleotide insertions and deletions are represented by black boxes. MHC-α (green), β2m (sand), Peptide (magenta), TCRα (teal), TCRβ (salmon), TCRα-CDR1 (red), TCRα-CDR2 (orange), TCRα-CDR3 (yellow), TCRβ-CDR1 (blue), TCRβ-CDR2 (green), TCRβ-CDR3 (maroon). (B–E) Histograms of V gene usage across the sequences of YLQ-, NYN-, PTD-, LLY-specific TCRs. Only the top ten of each gene are shown.
4.1 Interindividual TCR repertoire influencing immune responsesIt is well well-established that TCR diversity declines with age. In individuals within the first two decades of life, TCRβ diversity in the naïve T cell repertoires is estimated to be 60-120 million, but a decline to 8-57 million is observed in individuals over 70 years of age (96, 97). This age-related decline in TCR repertoire has been confirmed in the antiviral responses to human influenza A viruses (98, 99). This age-related decline in TCR repertoire has been observed in the antiviral responses to human influenza A viruses (98, 99). The numbers of antigen-specific CD8+ T cells across universal influenza epitopes were reduced in the elderly, although their effect/memory phenotype remained stable (98). Interestingly, the mortality rate of elderly COVID-19 patients is notably higher than that of young and middle-aged patients, while children mostly exhibit milder disease outcomes when infected with SARS-CoV-2 (2, 100, 101). Further studies are needed to clarify whether aged and less diverse TCR repertoires impact the ability of elderly patients to generate sufficiently robust T cell response to SARS-CoV-2. HLA variation is another factor of concern, influencing the composition of TCR genes by affecting both intra-thymus and extra-thymus clonal selection (102). A study by Francis et al. demonstrated that HLA variations significantly affect the CD8+ T cell repertoire shape and utilization of immune recall upon SARS-CoV-2 infection (84). Genetic differences in the HLA genes directly affect the binding affinity of MHC molecules to antigens, which in turn confer susceptibility or resistance to viral infections (32, 33, 103).
4.2 Diversity of TCR repertoire after SARS-CoV-2 infection and vaccinationThe size, frequency, and publicity of individual clonotypes within the TCR repertoire can provide insights into both successful and failed immune responses. During SARS-CoV-2 infection, the diversity and clonability of TCR repertoire peaked within 8-14 days (104, 105), and then returned to base levels within a week after virus elimination (106). In COVID-19 convalescent individuals, SARS-CoV-2 peptide continue to mediate long-term immune responses, with robust functional T cell responses persisting for up to 6 months post-infection (107). A study by Cohen et al. evaluated 254 COVID-19 patients longitudinally up to 8 months and found that virus-specific CD4+ and CD8+ T cells were polyfunctional and maintained with an estimated half-life of 200 days (108). Long-term immunity against SARS-CoV-2 is primarily driven by the clonal diversity of antigen-specific T cell responses (107, 109). Studies have demonstrated that highly diverse TCR repertoires can offer protection against a variety of antigens, including those from CMV, EBV, and HIV (110, 111), and such repertoires may be associated with a higher level of affinity, affinity, and overall functionality in immune responses (112, 113).
Vaccination against SARS-CoV-2 infection elicit strong T cell responses. Vaccine-induced CTL expansion seems to be relatively weak and results in fewer distinct clonotype clusters compared to CTLs induced by natural infection (114). Despite the rapid contraction of the circulating T Cell responses to SARS-CoV-2 mRNA vaccination, there is a persistent memory that were readily detectable in most individuals out to 235 days after vaccination (115). Repeated mRNA vaccination lead to large expansions of memory spike-reactive T cell clonotypes, most of which were CD8+ T cells, while also eliciting diverse spike-reactive T cell clonotypes not observed before vaccination (116). Infection-induced spike-specific CD8+ T cell memory plays an important role in the formation of circulating T cell bank size and clonal composition after vaccination (117), and mRNA vaccination promotes the expansion of memory CD8+ T cells (117, 118). As both virus- and vaccine-induced antigen-specific TCR repertoires undergo significant clonal contraction over time, coupled with an overall decline in immune response, booster vaccination may be the primary strategy to enhance long-term protection (109, 119).
Viral infection triggers massive T cells that can recognize specific antigens, resulting in skewing of the TCR repertoire toward these antigen-specific T cells (120). It has been demonstrated that certain V, D, and J fragments are over-expressed or under-expressed in COVID-19 patients with different clinical pictures (121). Compared with symptomatic patients, asymptomatic patients exhibit an overrepresentation of some TCR genes, including TRAV (AV17, AV12-1, AV19, AV35, and AV41), TRBV (BV12-5 and BV19), TRAJ16, and TRBJ2-1 (122, 123). Symptomatic patients, on the other hand, have higher frequencies of TRAV2, AJ8, AJ40, BV3-1, and BV5-1 (122). Severe patients tend to highly express TRBV5-6, BV14, BV13 and BV24-1 (124). Furthermore, a study showed that 25 sequences within the central parts of CDR3 region could be used to predict severe infection, emphasizing the significant impact of distinct clonal expansion on disease progression (125).
4.3 SARS-CoV-2 CTL epitopes recognized by public and private TCRsInterestingly, despite an estimated potential TCR diversity of 1015 (126, 127), TCRs that recognize a common ligand typically exhibit convergent sequence features in CDR3 residues that directly contact the peptide (111, 128), as well as CDR1 and CDR2 residues that can also contact the peptide and MHC, which are also known as “public” TCR motifs (129). In previous HIV-related studies, viral control has been linked to the presence of more cross-reactive public TCR clones, which may play a role in limiting viral escape pathways (130, 131). Several studies have identified “public” TCR in COVID-19 convalescents, characterized by conserved CDR motifs within and between individuals (71, 104, 132, 133). Ford et al. identified public CD8+ and CD4+ TCR motifs associated with SARS-CoV-2 spike specificity through TCR sequence similarity clustering (116). Analysis of over 4,000 epitope-specific TCR sequences showed that all SARS-CoV-2 exposures elicit diverse repertoires characterized by shared TCR motifs, confirmed by monoclonal TCR characterization (134). Here, we summarized TCR repertoires for 22 epitopes (each with a gene count >30) from the public VDJ database (135), as shown in Table 2. Consistent with previous studies (71, 133, 136), we found that TRAV12-1 (53%, 462/872) and TRBV7-9 (18%, 196/1110) were used by most YLQ-specific TCRs (Figure 2B). Likewise, TCRs recognizing other SARS-CoV-2 epitopes (e.g., NYN, LLY, PTD) display notable bias in the usage of specific TCR gene fragments (Figures 2C–E). For example, TRAV12-1 gene was commonly employed by YLQ, NYN, and PTD specific TCRs. Half of the epitopes we counted, such as SPR, TTD, QYI, LTD, RLQ, NYN, FTS, PTD, VYF, RVA, and WPV were dominated by the TRBJ2-7 gene (Table 2). This strong bias of TCR gene usage among epitope-dependent TCRs likely highlights the significance of germline-encoded features in TCR recognition, as previously reported in other antiviral immune responses (137, 138).
In addition to public TCRs, the TCR repertoire generated in response to a specific epitope varies between individuals, often referred to as “private” responses. The RLQ-epitope (spike1000-1008, RLQSLQTYV) is another immunodominant epitopes located on the SARS-CoV-2 spike protein, triggering cellular responses in most HLA-A*02:01+ convalescents (71). Unlike the highly “public” TCRs generated in response to the YLQ-epitope (71), TCRs responding to RLQ epitopes tend to be predominantly “private” and exhibit greater diversity (Table 2). The recognition of A*02-RLQ by receptors with diverse CDR3α and CDR3β pairings diminishes the publicity of A*02-RLQ responses between individuals, enabling them to recognize MHC and peptide in a manner that reduces the likelihood of identical or very similar V(D)J rearrangements in different individuals (28).
4.4 Cross-reactiveness of T cell repertoire in human coronavirusBased on the genomic similarities between SARS-CoV-2 and HCoVs, cross-reactive T cells may underlie the extensive heterogeneity observed in COVID-19 disease. SARS-CoV-2 T cell reactivity was mostly associated with CD4+ T cells, with a smaller contribution by CD8+ T cells (9, 10, 139, 140). Nevertheless, some reports provide the basis for a limited representation of cross-reactive CD8+ T cell responses (19, 60, 84, 85, 141). Minervina et al. detected certain T cell clones in memory fragments at pre-infection time points, suggesting participation of pre-existing cross-reactive memory T cells in the immune response to SARS-CoV-2 (19). Francis et al. reported that the clonal diversity of T cell responses to HLA-B*07:02 allele correlates with pre-existing immunity, allowing efficient presentation of homologous epitopes from both SARS-CoV-2 and HCoVs (84). Cytotoxic T cell responses against SPR-HLA-B*07:02 (N105-113, SPRWYFYYL) are often associated with mild cases of COVID-19 (80, 84, 142–145). Augusto et al., found that T cells from pre-pandemic samples from individuals carrying HLA-B*15:01 were reactive to the immunodominant SARS-CoV-2 spike-derived NQK epitope (60). Additionally, Shimizu et al. found that CD8+ T cells in response to a selected dominant QYI epitope display multifunctionality and cross-functionality across HCoVs in HLA-A24+ donors (141).
The HLA-B*07:02-restricted SPR epitope is one of the most cross-reactive epitopes, showing a high frequency in unexposed pre-pandemic samples (80, 145). Notably, the SPR peptide sequence is identical in SARS-CoV-2 and SARS-CoV-1, differing by only one residue in OC43-CoV-1 and HKU-1-CoV-1 (LPR), three residues in 229E (SPK) and four in NL63-CoV-1 virus (PPK), as shown in Figure 3A. Lineburg et al. reported that CD8+ T cells exhibit cross-reactivity between SARS-CoV-2 SPR and OC43/HKU-1-derived LPR peptide, but CD8+ T cells were unable to cross-recognize SPK and PPK peptides (85). The crystal structures of SPR-HLA-B*07:02 and SPK-HLA-B*07:02 elucidate these outcomes. Despite sharing common motifs in P1-2 (SP) and P6-9 (FYYL), differences in peptide sequences at P3-5 result in distinct conformations for SPK and SPR (Figure 3B). In the structure of SPR-HLA-B*07:02, four of the five aromatic residues create an interaction network, forming a compact and substantial binding surface for potential interaction with TCRs (Figure 3C). In comparison, the SPK peptide predominantly exposes the three carboxyl residues (P6-8) at the C-terminus (Figure 3C). Due to their high sequence identity, the LPR peptide (from OC43 and HKU-1) may adopt a conformation similar to that of SPR peptide (found in SARS-CoV-1 and SARS-CoV-2), providing the foundation for cross-reactivity among CD8+ T cells in HLA-B7+ individuals. However, despite sharing >55% (5/9) sequence identity, the distinctive structures account for the relatively low-level CD8+ T cell cross-reactivity observed between these peptides.
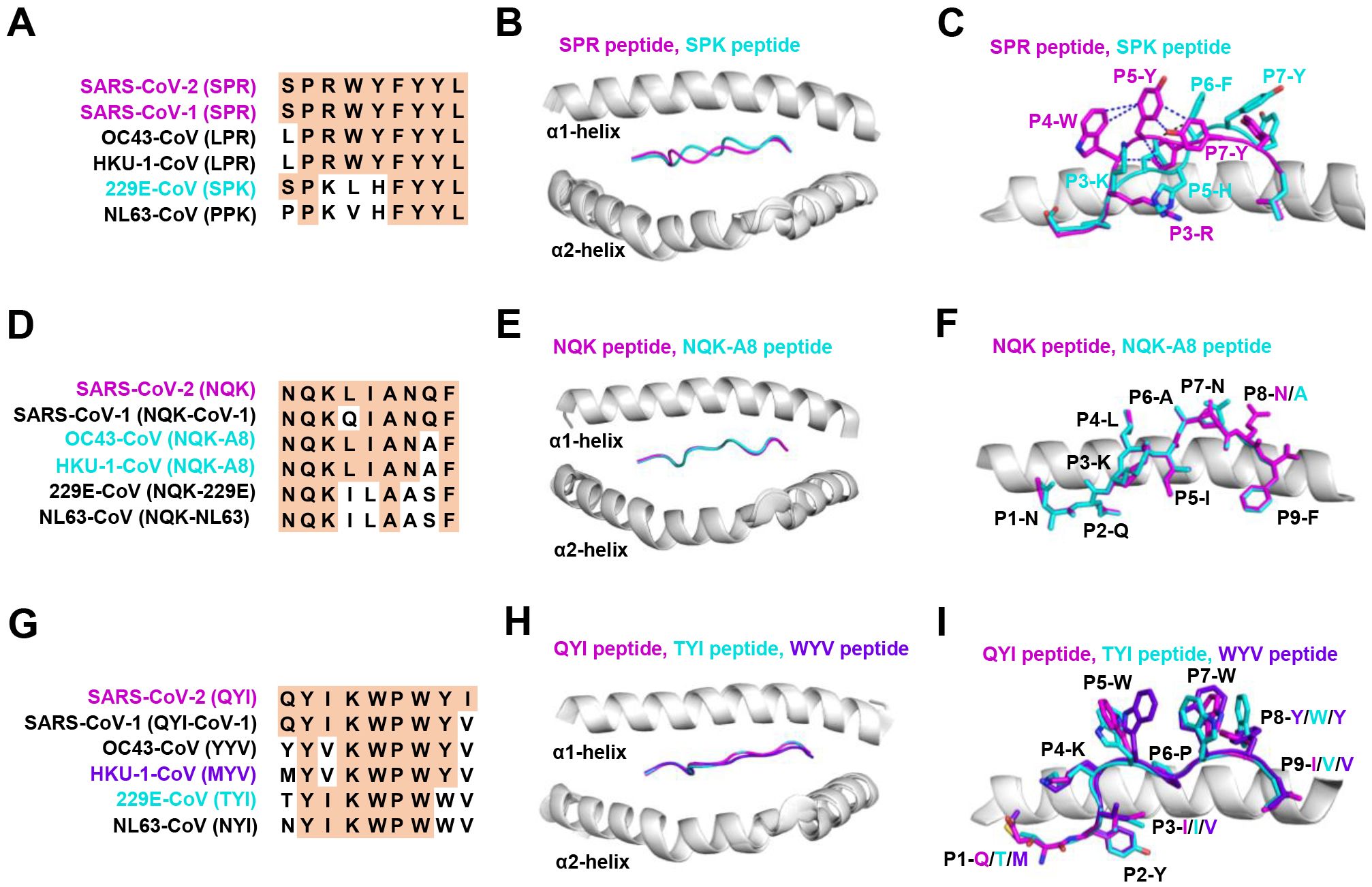
Figure 3. The structural basis of SPR, NQK, and QYI peptides for selective T cell cross-reactivity. (A) Peptide homologs of other HCoVs compared to the SARS-CoV-2 SPR peptide. (B) Structural superposition of the SPR-HLA-B7 (ID 7LGD) and SPK-HLA-B7 (ID 7LGT). SPR peptide (magenta), SPK peptide (cyan), HLA-B7 (grey). (C) Top view of the SPR-HLA-B7 and SPK-HLA-B7, with stick representation of the SPR and SPK peptides. Blue and black dashed lines indicate intra-peptide interactions of the SPR and SPK peptide, respectively. (D) Peptide homologs of other HCoVs compared to the SARS-CoV-2 NQK peptide. (E) Structural superposition of the NQK-HLA-B15 (ID 8ELH) and NQK-A8-HLA-B15 (ID 8ELG). NQK peptide (magenta), NQK-A8 peptide (cyan), HLA-B15 (grey). (F) Top view of the SPR-HLA-B7 and SPK-HLA-B7, with stick representation of the SPR and SPK peptides. (G) Peptide homologs of other HCoVs compared to the SARS-CoV-2 QYI peptide. (H) Structural superposition of the QYI-HLA-A24 (ID 7EJL), TYI-HLA-A24 (ID 7EJM), and MYV-HLA-A24 (ID 7EJN). QYI peptide (magenta), TYI peptide (cyan), MYV peptide (purple blue), HLA-A24 (grey). (I) Top view of the QYI-HLA-A24, TYI-HLA-A24, and MYV-HLA-A24, with stick representations of the QYI, TYI and MYV peptides.
Most NQK-specific reactive T cells display a memory phenotype, exhibit highly polyfunctional, and cross-react to a peptide from seasonal coronaviruses (60, 61). The NQK peptide of SARS-CoV-2 differs by only one residue in SARS-CoV-1, OC43-CoV-1, and HKU-1-CoV-1, and by three residues in 229E-CoV-1 and NL63-CoV-1 virus (Figure 3D). Crystal structures of peptide-HLA-B15 complexes reveal that the peptides NQKLIANQF (from SARS-CoV-1) and NQKLIANAF (NQK-A8, from OC43-CoV and HKU1-CoV) have similar stabilization properties (Figures 3E, F). This structural similarity of the peptides underpins T cell cross-reactivity of high-affinity public T cell receptors, providing a molecular basis for HLA-B*15:01-mediated pre-existing immunity (60). A similar pattern of peptide binding to HLA was observed with another dominant epitope, HLA-A24 restricted QYI (141). The QYI exhibits high sequence homology with SARS-CoV-1, OC43 (YYV), HKU-1-CoV-1 (MYV), 229E-CoV-1 (TYI), and NL63-CoV-1 (NYI) (Figure 3G). The cross-reactivity of the QYI epitope depends on the structural pattern of the peptide-HLA-A*24:02 complex and the combinations of TCR sequences (141). The structures of QYI-HLA-A24, TYI-HLA-A24, and MYV-HLA-A24 are almost identical, with rmsd (root mean square deviation) of 0.15 Å (QYI and TYI), 0.41 Å (QYI and MYV), and 0.41 Å (TYI and MYV), respectively (Figure 3H). The side chains of the peptides at P1, P4, P5, P7 and P8 are directed to the solvent region, and their structural orientations are slightly different (Figure 3I). This particular region forms a raised structure and is expected to interact directly with TCRs by their side chains. It is reasonable to speculate that some CD8+ T cells targeting seasonal coronaviruses may exist as long-term memory cells within the population. If these cross-reactive T cells are stimulated by COVID-19 vaccine or viral antigen, they could be skewed toward SARS-CoV-2.
5 CD8 + T cell immune escape by SARS-CoV-2 variants5.1 Antigenic mutations and loss of CTL epitope-specific responsesGiven the active role of T cells in combating viral infection, some mutations could potentially lead to the loss of CTL epitopes that evade recognition by CD8+ T cells (Table 3). Mutations within CD8+ epitopes in S protein (YLQ-L270F, LLF-L822F, and SII-S691P/S691C/I692T), M protein (GLM-L90F), N protein (MEV-M332I/L331S/L331F) were noted in one study during the course of acute infections, resulting in loss of epitope-specific responses (20). Prolonged SARS-CoV-2 infection in immunocompromised hosts may create a great opportunity for T cell escape. For instance, in the case of chronic SARS-CoV-2 infection, the NSP3 T504P mutation has been reported to result in the loss of CTL response (90, 146). These findings are limited to a few cases and suggest the need for more prospective cohort studies to systematically assess the risk of T cell escape in certain patient populations.
As the T cell responses target epitopes across the SARS-CoV-2 genome, the footprint of T cell escape is more broadly distributed than antibody-driven changes. In multiple SARS-CoV-2 lineages, some mutations within the immunodominant ORF1a (TTD- T1637I, T1638I, P1640S, P1640L, P1640H) ORF3a (FTS-Q213K) and N protein (QRN-P13L/S/T, KTF-T362I, P365S) CD8+ T cell epitopes resulted in a complete loss of recognition (88). Among these mutations, the N protein P13L mutation is present in Omicron within B*27:05-restricted CD8+ epitopes. Given the hypothesis that VOCs arise in chronic infections, it is tempting to speculate that the presence of P13L in Omicron reflects selection due to T cell stress during chronic infection, in addition to the constellation of spike mutations that are likely driven by antibody pressure. The mutant YLQ-P272L epitope could not be recognized by over 120 YLQ-specific TCRs, which may allow viral variants to escape from vaccine-induced T cell responses (28, 29). Spike-encoded L452R and Y453F led to the loss of HLA-A24-restricted CTL responses (91). In addition to evading antibodies and enhancing ACE2 binding affinity (147), the role of T cells in driving this change is uncertain. The extent to which these observations also represent incidental effects of mutations driven by other stresses on T cell responses is currently unknown.
5.2 Mechanisms of CTL immune escape by SARS-CoV-2 variantsT cell escape can occur through several mechanisms. Amino acid changes within epitopes or flanking regions can disrupt antigen processing (148), and changes to anchor residues can interfere with MHC/TCR binding to epitopes (29, 92, 149). Both these mechanisms can result in irreversible loss of T cell responsiveness to a particular epitope. To better understand the mechanism of CTL immune escape achieved by alterations in the binding between ligands and receptors, researchers have resolved several crystal structures of TCR-pHLA ternary complex and pHLAs loaded with original or mutant SARS-CoV-2 peptides, including YLQ, YLQ-P272L, RLQ, RLQ-T1006I, NYN, NYN-Y453F, KIA, KIA-K417T peptides (Table 4). All of eight TCR-pMHC ternary complexes loaded with SARS-CoV-2 peptides are symmetrically docked on pHLAs in a canonical diagonal orientation (Figure 4).
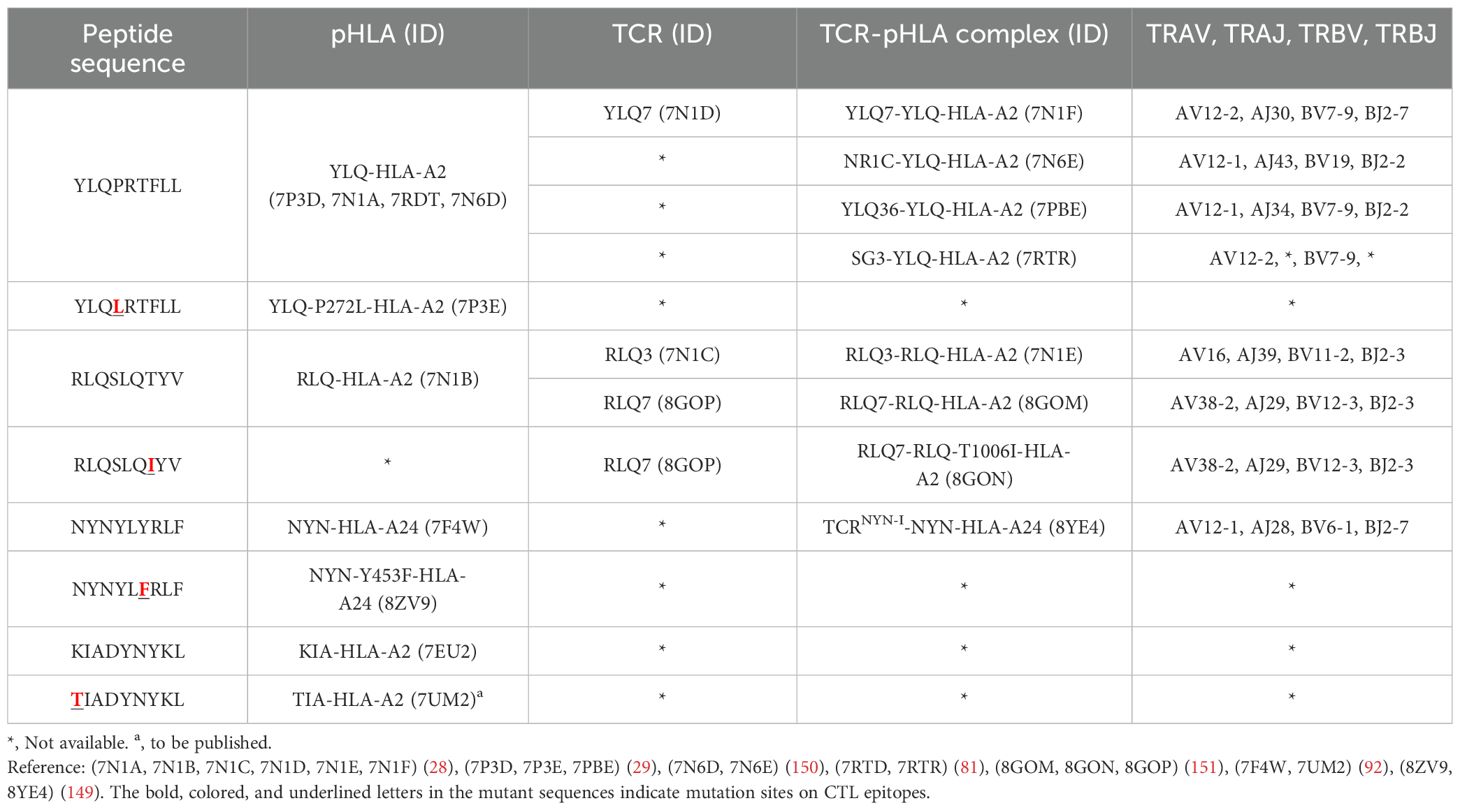
Table 4. Published crystal structures of TCR-pHLA and pHLA associated with SARS-CoV-2 CTL immune escape.
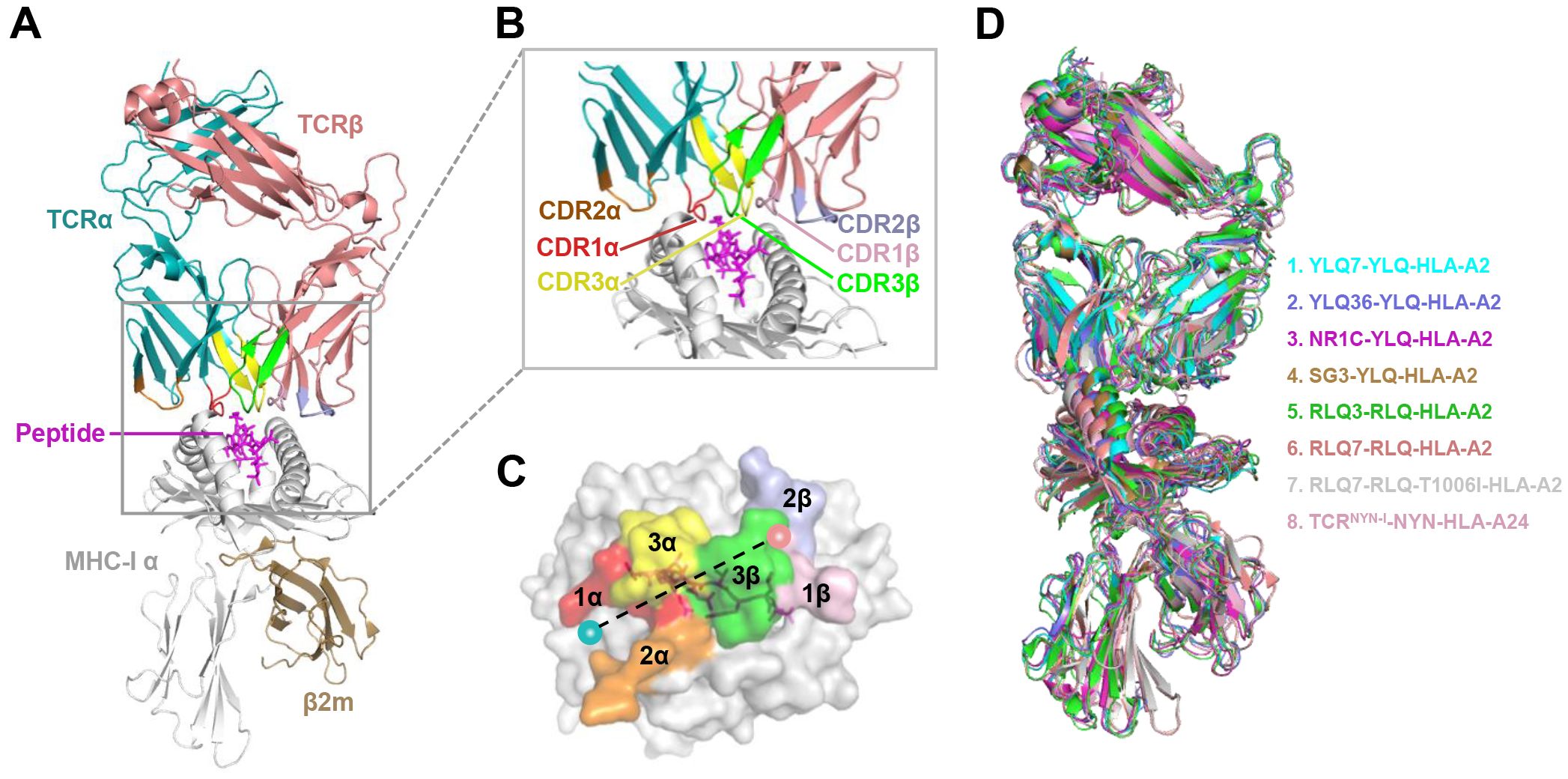
Figure 4. Schematic view of the TCR-pMHC I ternary complex. (A, B) Side view of YLQ7-YLQ-HLA-A2 (ID 7N1F). MHC-α (grey), β2m (sand), YLQPRTFLL-peptide (magenta), TCRα (teal), TCRβ (salmon), CDR1α (red), CDR2α (orange), CDR3α (yellow), CDR1β (light pink), CDR2β (light blue), CDR3β (green). (C) Footprint of TCR YLQ7 on YLQ-HLA-A2. (D) Cartoon comparison of eight TCR-pMHC ternary complexes presenting SARS-CoV-2 CD8+ T cell epitopes, including YLQ7-YLQ-HLA-A2 (ID 7N1F, cyan), YLQ36-YLQ-HLA-A2 (ID 7PBE, slate), NR1C-YLQ-HLA-A2 (ID 7N6E, magenta), SG3-YLQ-HLA-A2 (ID 7RTR, sand), RLQ3-RLQ-HLA-A2 (ID 7N1E, green), RLQ7-RLQ-HLA-A2 (ID 8GOM, salmon), RLQ7-RLQ-T1006I-HLA-A2 (ID 8GON, grey), TCRNYN-I-NYN-HLA-A24 (ID 8YE4, light pink).
Mutations within the epitope may destabilize or reassemble the pHLA complexes (20, 92). For example, our previous study showed that the K417N and Y144del mutations lead to failure in the formation of the KIA-HLA-A2 and GVY-HLA-A11 complexes, respectively, blocking the first step in antigen presentation (92). Additionally, structural analysis shows that the positively charged side chain of N-terminal lysine forms a π-cation interaction with the indole ring of W167 of HLA-A2, and K417N/T mutations at this position (K417T) are expected to abolish the π-cation interaction at this position (92). The loss of this key peptide-HLA interactions may provide SARS-CoV-2 variants with an opportunity to evade cellular immunity.
Mutations within the epitope may disturb or change the peptide-dependent TCR contacts (29, 149). For the dominant YLQ epitopes, the original YLQ peptide forms six intra-peptide bonds to stabilize TCR binding, while the mutant YLQ-P272L-HLA-A2 (YLQLRTFLL) structure exhibits fewer internal contacts (Figure 5A). The P5 arginine of the peptide underwent a significant conformational shift during the binding of TCR YLQ36 (Figure 5B). Superimposed structures of YLQ-P272L-HLA-A2 and YLQ36-YLQ-HLA2 complex revealed that the leucine of YLQ-P272L might protrude within 1˚ of the YLQ36 CDR3α loop, creating a steric hindrance between them (Figure 5C). This steric hindrance could potentially jeopardize the interaction between CDR3α and YLQ peptide, resulting in the loss of YLQ36 T cell recognition. Similarly, the spik-Y453F mutation within the NYN epitope is another dominant mutation that triggers HLA-A24-restricted CTL immune escape (91). In order to explore the escape molecular mechanism, we determined the crystal structures of original NYN-HLA-A24 (NYNYLYRLF) (92), mutant NYN-Y453F-HLA-A24 (NYNYLFRLF), and a ternary structure of TCRNYN-I-NYN-HLA-A24 (149). Structural analysis showed that after mutation or TCRNYN-I binding, the conformation of the NYN peptide changed significantly, especially P4-Tyr, P5-Leu, and P6-Tyr/Phe (Figures 5D, E). The hydrophobic phenylalanine in the SARS-CoV-2 variants may disrupt contact network of the original tyrosine with TCRNYN-I, suggesting that despite competent presentation by HLA, the mutant Y453F peptide failed to establish a stable TCR-pHLA ternary complex due to reduced peptide: TCR contacts(Figure 5F). Unlike the P272L and Y453F dominant mutations, the RLQ-T1006I mutation was found to be tolerated in HLA-A24-restricted CTL activation (28, 30, 151). The T1006I substitution leads to a structural rearrangement of the HLA peptide-binding groove and alters the conformation of peptide residues P3-Gln and P6-Gln (Figure 5G). TCR RLQ7 CDR1α engages the N-terminal region of the original RLQ peptide via two direct and three water-mediated hydrogen bonds (Figure 5H), while TCR RLQ7 forms two new compensating hydrogen bonds with mutant RLQ-T1006I peptide (Asp31α with P4-Ser, Asp31α with P5-Leu) and an additional hydrogen bond with HLA-A2 (Glu29α with Arg66-HLA-A2) (Figure 5I). Similar stabilities of the two complexes may provide a reasonable explanation for the limited CTL immune escape.
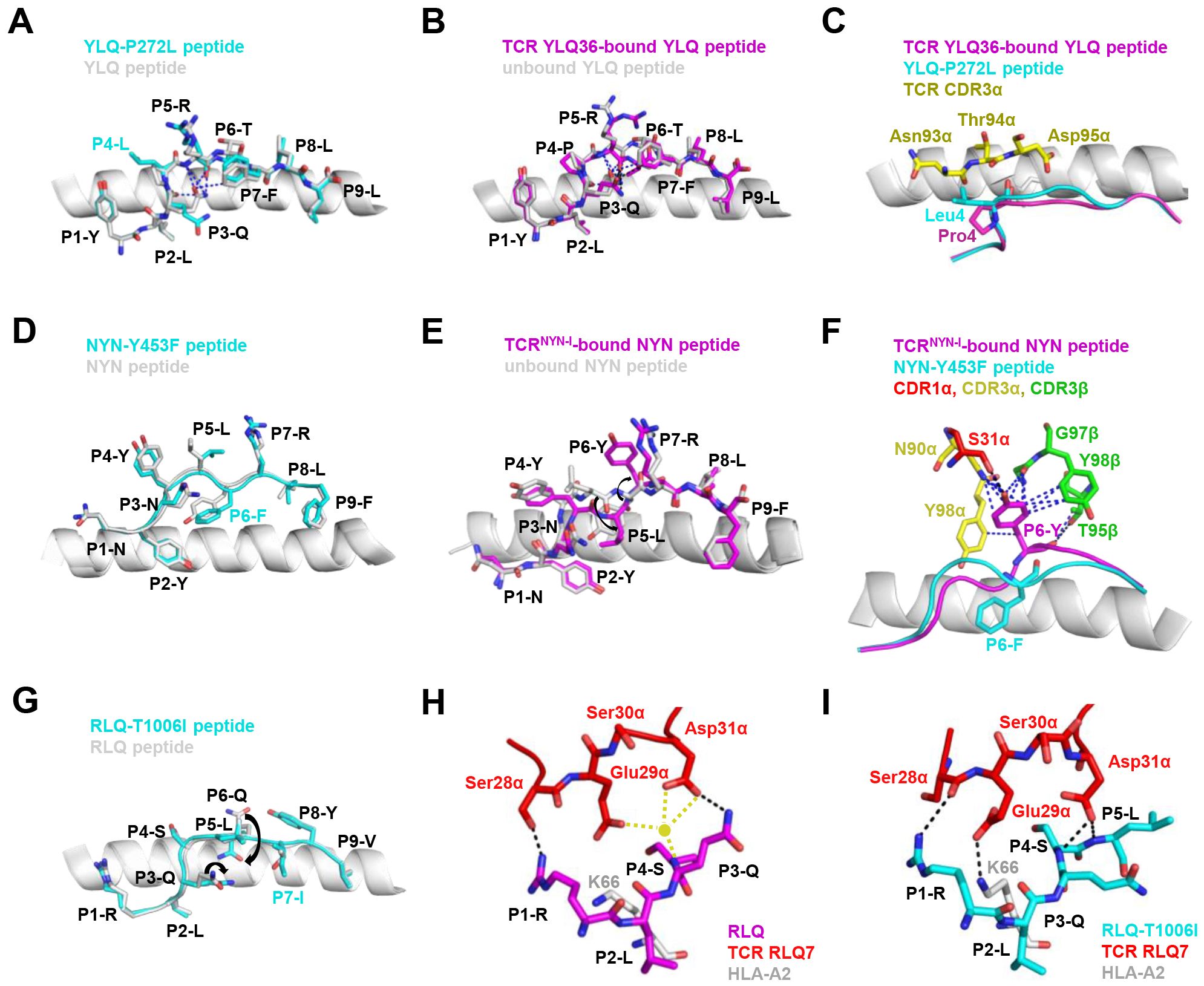
Figure 5. Mechanism of CTL immune evasion mediated by SARS-CoV-2 epitope mutation. (A) Comparison of YLQ-HLA-A2 (ID 7P3D) and YLQ-P272L-HLA-A2 (ID 7P3E). Intrapeptide bonds present in YLQ-HLA-A2 are shown as blue dashes. YLQ peptide (grey sticks), YLQ-P272L peptide (cyan sticks), HLA-A2 (grey cartoon). (B) Comparison of unbound YLQ-HLA-A2 (ID 7P3D) and TCR-bound YLQ-HLA-A2 (ID 7PBE) peptide presentation. Intrapeptide bonds present in TCR-unbound and TCR-bound YLQ-HLA-A2 are shown as blue and black dashes, respectively. TCR-bound YLQ peptide (magenta), TCR-unbound YLQ peptide (grey sticks), HLA-A2 (grey cartoon). (C) YLQ and YLQ-P272L P4 residues shown as magenta and grey sticks, respectively. YLQ36 CDR3α loop shown as yellow sticks. (D) Comparison of NYN-HLA-A24 (ID 7F4W) and NYN-Y453F-HLA-A24 (ID 8ZV9). NYN peptide (magenta), NYN-Y453F peptide (cyan), HLA-A24 (grey). (E) Comparison of unbound NYN-HLA-A24 (ID 7F4W) and TCR-bound TCRNYN-I-NYN-HLA-A24 (ID 8YE4) peptide presentation. The van der Waals and hydrogen bonds between P6-Tyr and TCRNYN are blue and black dashes, respectively. (F) NYNYLYRLF and NYNYLFRLF P6 residues shown as magenta and cyan sticks, respectively. HLA-A24 shown as grey cartoon. TCRNYN-I CDR1α, CDR3α and CDR3β loops shown as red, yellow and green sticks, respectively. Hydrogen bonds are shown in black dashes. Van der Waals contacts are shown as blue dashes. (G) Structural rearrangements in RLQ-HLAs resulting from the T1006I mutation. RLQ-HLA-A2 (TCR RLQ7-HLA-A2, ID 8GOM), RLQ-T1006I-HLA-A2 (TCR RLQ7-T1006I-HLA-A2, ID 8GON), RLQ peptide (magenta), RLQ-T1006I peptide (cyan), HLA-A2 (grey). (H) Interactions between CDR1α of RLQ7 (red) and the RLQ peptide (magenta). Yellow sphere indicates an interfacial water molecule. Water-mediated hydrogen bonds are yellow dashed lines. Hydrogen bonds are black dashed lines. (I) Interactions between CDR1α of RLQ7 (red) and the RLQ-T1006I peptide (cyan). Hydrogen bonds are represented by black dashed.
Disruption or inhibition of antigen processing by mutation is a third mechanism of T cell immune escape. The components of the antigen processing pathway have preferences for their optimal amino acid sequences (152). It has been demonstrated in HIV-related studies that differences in amino acid sequence in the region flanking the epitope impaired the intracellular processing and presentation of epitope (153, 154). In the context of SARS-CoV-2 infection, a study proposed that variants may disrupt the HLA-I antigen presentation pathway by depleting proteasomes and altering the activity of ubiquitination enzymes, thereby preventing infected cells from presenting antigen proteins (148). A deeper understanding of the mechanisms by which SARS-CoV-2 evades host immune surveillance through the ubiquitin-proteasome system and HLA-I presentation warrants further investigation.
6 ConclusionT cells are the backbone of the immune system and play a crucial role in the progression of COVID-19. HLA allele-related studies are essential for assessing the role of HLA in the immune response against SARS-CoV-2. Although some studies have demonstrated that HLA alleles were associated with differential susceptibility, these results were not consistent across studies. The lack of large-scale HLA typing in samples has limited the scope of research, with most studies involving sample sizes fewer than 190. Future large-scale studies are needed to provide a comprehensive understanding of the association between HLA genotypes and the evolving SARS-CoV-2 variants, thus improving our understanding of the association between antigen presentation and disease progression. Moreover, T cells carry a natural “barcode” sequence in their variable TCR region, specifically in the CDR3 component. Gene mutations and recombination endow the TCR repertoires great diversity and poly-clonality. Studies related to TCR-seq provide evidence for the close relationship between TCR diversity and anti-viral immunity. In the context of viral infection, the preferential selection of T cell clones narrows the TCR repertoire for antigen selection. Nevertheless, the full influence of SARS-CoV-2 on the TCR repertoire remains to be evaluated.
Although the characteristics of HLA polymorphisms and TCR repertoire in SARS-CoV-2 infection have been initially elucidated, the emergence of immune evasion variants complicates a comprehensive understanding of these relationships. Since the onset of the pandemic, SARS-CoV-2 has continued to evolve and adapt to its host, remaining a relatively new coronavirus. These variants are more prone to cause immune escape and vaccine escape. Studying the mechanisms of immune and vaccine escape remains a significant challenge. There is a need to conduct ongoing assessment of vaccine efficacy against these evolving variants, which may contribute to elucidate the drivers of the spread and evolutionary success of circulating variants. Addressing these unresolved issues promptly is crucial to ending the current pandemic and preparing for potential future ones.
Author contributionsTJ: Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing – review & editing. SD: Conceptualization, Data curation, Formal analysis, Resources, Software, Writing – original draft, Writing – review & editing. ZX: Formal analysis, Supervision, Visualization, Writing – review & editing. JH: Formal analysis, Writing – review & editing. YY: Formal analysis, Writing – review & editing. ZL: Formal analysis, Writing – review & editing. HZ: Formal analysis, Writing – review & editing. SW: Formal analysis, Writing – review & editing. FZ: Formal analysis, Writing – review
留言 (0)