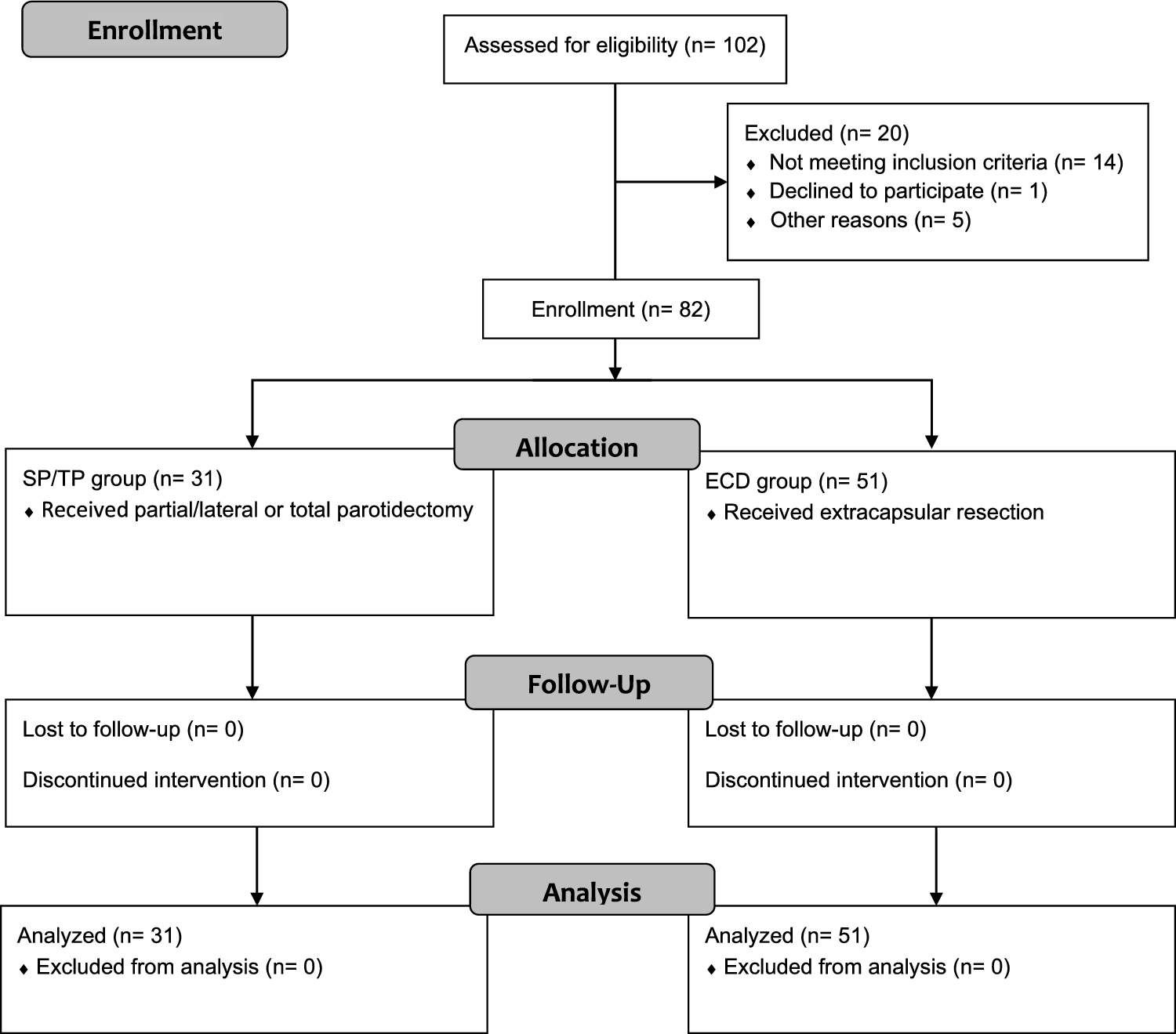
This prospective clinical study was approved by the Ethics Committee of the xxx (approval number 374_18 B). It was conducted according to the Declaration of Helsinki and followed the STROBES guidelines [18] as well as the guidelines of the “Quality Improvement in Postoperative Pain Treatment” (QUIPS) registry [19]. The study was registered in the German Clinical Trial Register (DRKS00016520). A total of 82 patients, who presented for elective salivary gland surgery at the xxx between March 29, 2021, and March 18, 2022, were included.
Inclusion criteria were defined as follows: surgery on the parotid gland due to a parotid mass irrespective of the clinically suspected dignity, written consent to data collection and processing, minimum age of 18 years, sufficient cognitive and verbal ability to understand the content of the questionnaires, an American Society of Anesthesiologists (ASA) status of I–III. and a body mass index (BMI) ≥ 19 kg/m2.
Patients were excluded who were undergoing revision surgery or radical parotidectomy, who did not provide written informed consent or who had undergone major skin resections with subsequent flap reconstruction or concomitant resections of the auditory canal or temporal bone. In addition, all patients who had not been treated postoperatively according to the standardized pain protocol of the Department of Otolaryngology were excluded from the statistical analysis.
The patient collective was divided into two groups. The first group (referred to below as SP/TP group) consisted of patients in whom the trunk of the facial nerve had been primarily exposed during surgery, i.e. superficial or total parotidectomy (n = 31). The second group (referred to below as ECD group) included all patients who had undergone extracapsular dissection (ECD) (n = 51). Assignment to the respective group was based on the surgical report. As the surgeons did not know before or during the operation whether the patient was a study participant, the choice of surgical technique was left to the surgeon’s discretion.
In all patients, anesthesia was induced according to the current department standard (fentanyl 1–2 µg/kg bodyweight (bw), propofol [TCI (target-controlled infusion) plasma level 4–8 ng/ml, Orchestra® Base Primea, Fresenius Kabi, Bad Homburg, Germany, plasma mode, Marsh mode), and rocuronium 0.3–0.5 mg/kg bw, intravenously (IV)]. To maintain anesthesia, plasma propofol TCI was reduced to levels between 2.5 and 4.0 ng/ml, and the strong intraoperative pain stimulus was treated with remifentanil 0.2–0.5 µg/kg/min run rate combined with fentanyl (1 µg × kg−1 bolus as required). To attenuate propofol injection pain, all patients received 40 mg lidocaine 2% IV before propofol administration.
Pain in the recovery room was treated with ibuprofen, acetaminophen, and metamizole as well as intravenous piritramide (0.05–0.2 mg × kg−1) as required up to a pain intensity of NRS (Numeric Rating Scale) < 3 to be transferred to a normal ward.
Patients on the ward were treated according to the standardized pain protocol of the Department of Otorhinolaryngology-Head and Neck Surgery, according to the WHO (World Health Organization) step-by-step scheme, if required and desired (see Supplement 1).
The standardized and validated QUIPS questionnaire ("Quality Improvement in Postoperative Pain Therapy") was used once a day during the first three postoperative days (PODs) to survey patients with regard to their pain, physical impairments, side effects of pain therapy and their general condition (so-called outcome parameters) [20]. This was supplemented by the so-called process parameters, filled out by the investigator, containing patient-relevant information, details on anesthesia and surgery, and analgesic consumption.
Moreover, all patients were screened for a neuropathic pain component using the validated 12-item painDETECT® questionnaire (PD-Q) (Pfizer Germany) on days 1 and 3. In this questionnaire, an overall score is calculated from all the patient's answers, resulting in a negative (0–12), indeterminate (13–18), or positive (19–38) screening result[21, 22].
During the daily visit patients were asked to fill out the questionnaires by themselves and were given assistance if desired. The same person performed the study inclusion, patient education, and the postoperative visits.
The documentation of demographic data as well as the completion of the QUIPS process parameter data sheet was performed by the same individual during and after the inpatient stay based on the digital and paper patient files. The data on the size of the resected tissue, measured in all 3 room levels, and the results of the histologic examination were taken from the histologic findings prepared by the Department of Pathology at the University Hospital Erlangen.
Primary endpoints were maximum and minimum pain as well as pain on ambulation (NRS 0 (“no pain”) – 10 (“worst pain imaginable”) on the first three postoperative days. Our main hypothesis was that the maximum pain on the first postoperative day would be greater in the SP/TP group. Secondary endpoints comprised a neuropathic pain component measured on POD 1 and 3 and analgesic consumption, measured on POD 1,2 and 3. Further secondary endpoints were perioperative and postoperative complications, treatment-related side-effects/pain-associated impairments, wish for more pain medication, and satisfaction with the pain therapy, measured on POD 1.
Statistical analysis
Metric variables were first checked for normal distribution using their histograms, QQ plots and Shapiro-Wilks tests.
To ensure good comparability, all metric variables were presented as median and 25th/75th percentile (Md [25th; 75th percentile]) and additionally as mean ± standard deviation (SD). Nominal variables were reported as absolute and relative frequencies [n (%)].
Independent t tests were calculated for normally distributed metric variables and Wilcoxon tests for non-normally distributed metric variables to compare the types of disaggregation. Nominal variables were tested for group differences using χ2 tests.
To test metric variables for group differences over time, robust mixed ANOVAs were performed with a 10% trim on both sides of the distribution with the between-subjects factor 'dissection type' and the within-subjects factor 'time'. This leads to the main effects (ME) "dissection type", "time" and the interaction "dissection type x time".
In the case of a significant interaction/a significant main effect, these were further differentiated in the follow-up using a robust t-test with a 10% cut-off on both sides.
For the independent t test, Cohen's d was calculated as the effect size, with d = 0.2 representing a small, 0.5 a medium and 0.8 a large effect. For the Wilcoxon test, rank-order correlation r and for the χ2 test phi/Cramer's V were reported, with r/phi/V of 0.1 representing a small, 0.3 a medium and 0.5 a large effect.
The effect size ξ2 is still reported for the effects of the robust ANOVA and for pairwise comparisons. This corresponds to the classical R2, with small, medium, and large effects, where 0.01 corresponds to a small, 0.09 to a medium and 0.25 to a large effect.
A priori power-analysis showed that a power of 0.80 could be achieved with 37 patients per group when a group difference of 1.0 and a standard deviation of 1.5 was assumed (alpha = 0.05). The actual sample size slightly differs due to practical circumstances.
The present analyses were conducted using R and the packages effectsize, effsize and WRS2 [23, 24].



留言 (0)