
記住我

The study will be conducted and reported according to Consolidated Standards of Reporting Trials (CONSORT) [36, 37] and extensions for non-pharmacologic treatment interventions and multi-arm parallel-group randomised trials and CONSORT-EHEALTH for improving and standardising evaluation reports of Web-based and mobile health interventions [38].
Study designThe trial design is a superiority parallel 2-arm individual-level single-blind Randomized Controlled Trial (RCT). All participants will be randomized into either receiving the self-help unguided self-help app incorporating CBT strategies targeting RNT in addition to usual practice or to receiving usual practice alone.
Potential participants for the trial provide initial consent to complete screening measures to determine if they are eligible to participate in the trial, i.e., showing elevated levels of RNT. Any potential participants who are found not to be eligible are automatically signposted to other sources of support. Once trial eligibility has been determined and consent to participate in the trial has been obtained, participants are individually selected at random (in a 1:1 ratio) to be offered the unguided self-help app or to continue with usual practice only.
Recruitment and study settingsWe seek to recruit 648 participants from within United Kingdom universities (focused on the partner universities in the grant: Exeter, Newcastle, Oxford, Cardiff, Southampton, and King’s College London, but also open to other UK universities). The recruitment strategy will include on-campus presentations, posters, stands, and presentations in lectures, online and website advertising; email to mailing lists; newsletters and other circulars and noticeboards within willing universities in the UK. A social media campaign will be designed and prepared to be carried out on different social networks (e.g., Facebook, Instagram, TikTok), including advertisements in social media.
Eligibility criteriaEligible participants will be: (1) over 16 years of age; (2) studying at a UK university; (3) able to provide informed consent; (4) having regular access to a smart phone (android or iOS), tablet, PC or laptop, necessary to use intervention; (5) available for the full duration of the trial (12 months); (6) able to complete consent and online questionnaires based on a basic literacy in English; (7) scoring above previously established cut-offs on standardized self-report measures of worry (Penn State Worry Questionnaire- Short-form, PSWQ-S) [39] and rumination (Ruminative Response Scale-Brooding, RRS-B) [40] , defined as scoring in the worst performing quarter on at least one of these measures, and scoring in the worst performing third on the second measure (in practice, this means scores of >11 for worst tercile, > 12 for worst quartile on the RRS-brooding scale and >24 for top tercile and >26 for top quartile on the Penn State Worry Questionnaire short-form). These thresholds are based on prior studies finding that individuals scoring in the worst quarter on measures of RNT have elevated risk for subsequent anxiety and depression [18, 19, 41].
Since this is a prevention trial, potential participants will be excluded at baseline if currently meeting diagnostic criteria for major depression (according to psychiatric DSM-V criteria), determined in structured self-report electronic screening using the LIDAS instrument [42]. Participants will also be excluded at baseline if presenting with highly elevated symptoms of depression indicating they require more specialist treatment. This is defined as having a score of 20 or higher on the Patient Health Questionnaire (PHQ-9) [43]. Other exclusion criteria include: active suicidality; any self-reported history of severe mental health problems such as bipolar disorder and psychosis and drug/alcohol dependence; and currently receiving psychological therapy, counselling or psychiatric medication including antidepressants.
Screening and consent procedurePotential participants who are interested in the study will be directed to our study website, (www.nurtureuniversity.co.uk) which provides further information. Interested participants can proceed directly to the Electronic Data Capture (EDC) system and undertake a brief pre-screener to check age and that the participant is a university student. If appropriate, the website visitor can watch a video version of the screening Participant Information Sheet (PIS) and/or read an electronic version of the PIS and an initial consent screen to provide contact details (email; mobile phone number), and to provide informed consent (screening consent form) to complete the screening questionnaires. After completing pre-screening, potential participants will be automatically emailed a copy of the screening PIS, privacy policy and completed screening consent form. Once this initial consent is provided, the participant will complete the screening assessment consisting of the LIDAS, PSWQ-S, RRS-B and PHQ-9 and if meeting eligibility criteria will be assigned to the trial (the screening process will in parallel be assessing participants for eligibility for a trial of internet cognitive behavioural therapy for students with elevated anxiety and depression).
Those meeting eligibility criteria following the screening assessment will then be asked to consent to take part in the trial after viewing a video version of the trial PIS and/or reading an electronic version of the trial PIS and completing an electronic trial consent form (trial consent form). Participants who provide consent are automatically emailed a copy of the trial PIS, privacy policy and completed trial consent form.
Individuals who are not suitable at pre-screen or screening (e.g., outside of age range, below threshold on RNT) will automatically be directed to a webpage explaining why they are not suitable for the trial. Those reporting mental health difficulties will be automatically guided to webpages providing information, guidance including to consult with their general practitioner (or equivalent), and weblinks and telephone numbers for help and support, including contact details for the trial team.
Baseline and follow-up assessmentsThe baseline assessment will take place after electronic informed consent to participate in the trial is provided and consists of web-based self-report measures to assess current and lifetime history of depression, current wellbeing, symptoms of anxiety and depression, social and work functioning, use of services and treatment received, academic grades, resilience, and stressful events, and levels of RNT (see outcome measures and Tables 1 and 2). The Lifetime Depression Assessment Self-report questionnaire (LIDAS) [42] will be used to assess lifetime major depression (MDD) diagnosis according to DSM criteria and is largely based on the widely used Composite International Diagnostic Interview (CIDI). It has been proven to be effective for determining history of depression through self-report in an online digital format, matching the needs for the current study [42]. It consists of a conditional sequence of pre-programmed questions assessing all the diagnostic criteria for depression, with logic cut-outs so that subsequent questions are determined by prior questions, keeping the assessment brief.
Table 1 Schedule of enrolment, interventions and assessments Table 2 Measurements and endpointsOnce consented participants have completed the baseline assessment, they will be randomised automatically by an independent computer system. Randomised participants will be informed of their randomisation allocation and signed up to use the relevant variant of the app via clinically trained members of the research team. The participant’s University email address is used for app set-up.
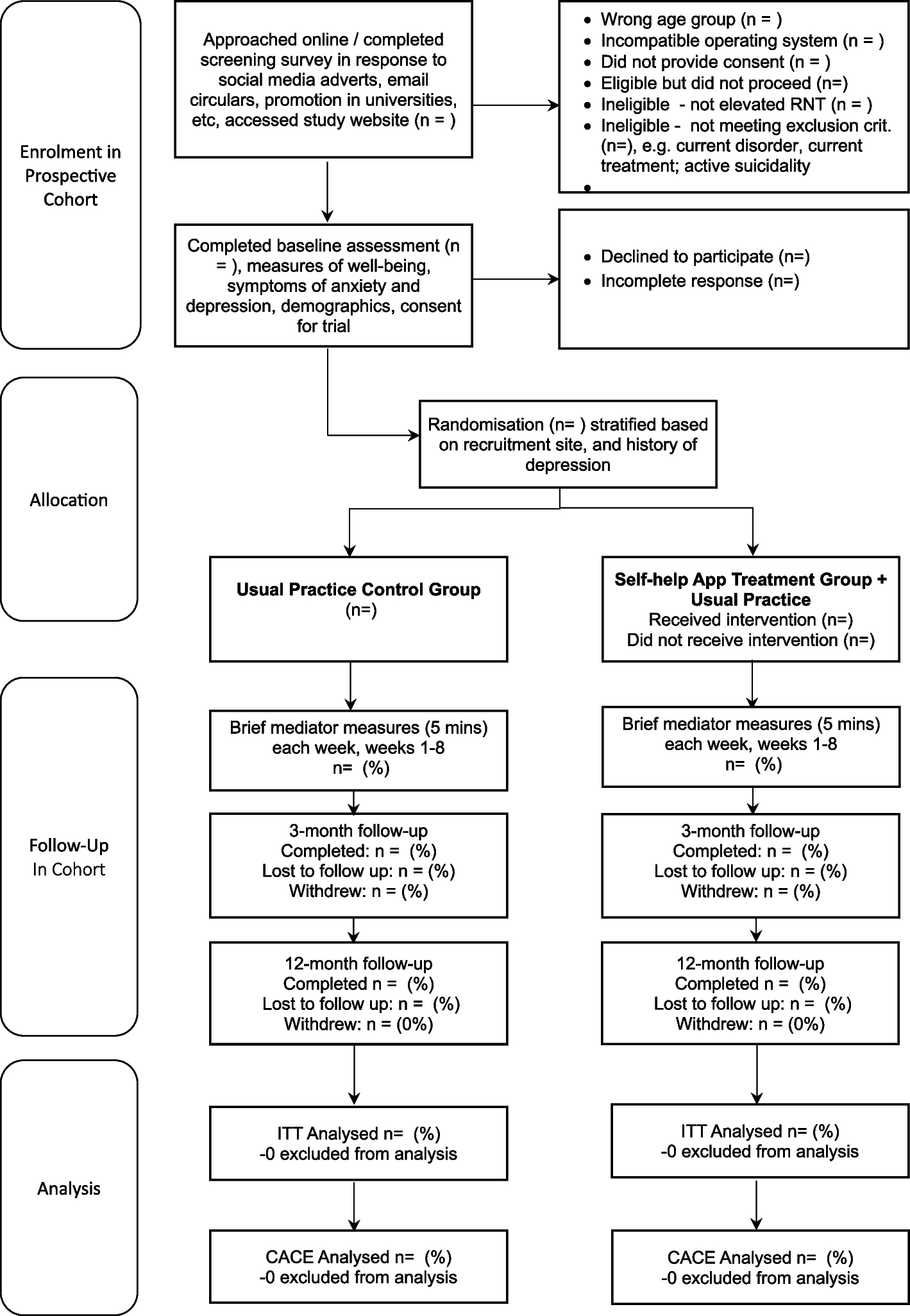
All participants will be followed up electronically at 3 months and 12 months post-randomisation. At each follow-up point, participants will be automatically sent emails and texts with links to enter their data into the EDC. Each assessment point will involve an automated weekly follow- up by email and then text and telephone follow-ups to participants who haven’t yet completed the EDC assessment at 3 months and 12 months. Tables 1 and 2 give an overview of all measurements. Figure 1 gives an overview of trial flow. Project researchers will be blind to treatment allocation but will be available to participants to answer queries about the trial or the assessments. Participants will receive honorariums for the completion of each of the brief mediator assessments occurring each week for 8 weeks post-randomisation, 3-month and 12-month follow-up assessments.
Fig. 1 Randomisation, intervention delivery and masking
Randomisation, intervention delivery and maskingParticipants will be randomised (in a 1:1 ratio) to the two intervention arms. Randomisation will be conducted automatically by means of a custom-built secure web service created and managed by the Exeter Clinical Trials Unit (ExeCTU), which interfaces with the trial database, and which will be independent of the trial researchers. To promote balance across key participant characteristics across intervention arms, randomisation will be stratified according to recruitment site (each of the six funded partner universities plus an “other” university category for students recruited elsewhere), and history of depression (no history vs past history of depression – given that this is a strong predictor of future depression and was used previously) [19]. Stratification will be used (on these two variables) and blocking will be used with minimum block size of 4 and maximum block size of 6, occurring at random to maintain concealment.
All the online recruitment and randomisation will be automated and independent of trial researchers. The Exeter CTU EDC system will contact an unblinded team member (administrator/therapist) indicating when an individual [by study ID] has been randomized to the active intervention. This team member will then access the relevant details in the EXCTU database and manually set up the participant in the mobile app intervention using administrator rights via the treatment platform dashboard. An email will also go to the participant indicating the condition to which they are randomised and informing them what to expect. The relevant unblinded team member will also monitor if the participant has accessed the intervention and check if any difficulties and encourage to sign-up.
All assessments will be routinely collected online using the EDC following automated reminders, without the involvement of researchers. Site researchers will be blind to treatment allocation. Site researchers will prompt all participants to complete follow ups by phone and text if they do not respond to the automated email reminders. Any unblinding in contact with a site researcher would be logged as a protocol deviation and only a researcher that remained blind will be able to prompt future follow-up from that participant.
InterventionsUsual practice controlThis control is what usual practice the participants may receive during the course of the 12-month follow-up. Usual practice, as received by the university student outside of the trial, may include no provision of intervention, local provision of intervention, support from their GP/family doctor, local health services or youth services, or provision of intervention within their university (e.g., well-being service; support and welfare staff). The nature of usual practice will be monitored and assessed by questionnaires at each follow-up assessment, determining what treatment and services participants have received since the last assessment.
Unguided RNT-focused self-help mobile app (experimental intervention group)The self-help app includes self-monitoring, psychoeducation, and active self-help exercises based on RNT-specific strategies from an evidence-based rumination-focused CBT intervention [35]. Core elements of the intervention are designed to break the ruminative habit and enable users to shift towards a more helpful processing style. This involves coaching participants to spot warning signs for rumination and worry, and then to plan alternative strategies. These include being more active, slowing things down, breaking tasks down, opposite action, relaxation, concrete thinking, becoming absorbed, self-compassion, and assertiveness. Participants are prompted to practice alternative strategies in response to their warning signs. The user interface includes text, pictures, audio-recordings, vignettes, animations, audio-exercises, quizzes, and questionnaires with tailored automated feedback. The app is entirely automated (i.e., self-guided) and designed for use on both iOS and Android phones. The app is accessed for free via each participant’s smartphone app store and is set up in the Minddistrict delivery platform. The app is structured in a logical order with participants needing to complete each module/lesson before proceeding to the next lesson. Each lesson can be completed within 5-30 mins and followed a structure of psychoeducation, student stories, experiential exercises, strategies to practice, making plans, and a quiz to test knowledge (see Table 3 for further detail).
Table 3 Detailed description of internet RNT-focused mobile app interventionThe content will focus on providing psychoeducation, tips, advice, strategies, and reflective exercises and learning tests relevant to reducing worry and rumination and building confidence. It has been adapted to be usable in a mobile app format and has been tailored through two iterations (one on paper with illustrative examples for a student focus group, one with n=6 students working through the app and providing feedback) to reflect student concerns, including specific sections on student issues and a series of diverse student vignettes illustrating different concerns and strategies through the app. The vignettes are designed to follow different student stories to illustrate different aspects of the intervention and different experiences of worry and rumination. The adaptation includes organising all information into “bite-sized” information that can be displayed on a mobile phone screen without the need to scroll down, typically with one key message per “page”, and with users then swiping forwards to the next “page” to make it user-friendly.
There is a “favourite” feature so that any exercise or video or information could be stored and accessed whenever the user wanted from their dashboard. The app also includes interactive features that the user can access whenever they want and as many times as they want, including features to complete diaries, set goals, and make plans.
Intervention adherenceThe use of the app will be assessed and recorded including number of times the app is used, for how long, and progress through the different modules. A minimum intervention dose for the app will be defined a priori, with “compliance” defined as completion of a pre-specified minimum level of usage of the unguided self-help intervention (at least 2 modules of 7 modules in treatment package). All participants in the usual practice control will be defined as receiving the minimum intervention dose.
OutcomesOutcomes will be assessed at baseline (pre-randomisation) and 3 months and 12 months post-randomisation.
Primary outcomeThe primary outcome measure will be the incidence of major depression over 12 months (primary endpoint), indexed by the LIDAS [42] assessing diagnosis retrospectively across the follow-up period.
Secondary outcomesSecondary outcomes include: the Patient Health Questionnaire-9 (PHQ-9), a well-validated measure of depression [43] and the Warwick -Edinburgh Mental Wellbeing Scale (WEMWBS) [44], a widely used and well-validated measure of wellbeing. The Generalized Anxiety Disorder-7 (GAD-7) questionnaire will be used to assess anxiety symptoms [45]. The Work and Social Adjustment Scale (WSAS) [46] will be used to measure functioning with respect to work/education, home management, social leisure, private leisure, and close relationships each rated from 0 not at all impaired to 8 severely impaired. A bespoke questionnaire will assess the use of university-based and non-university-based healthcare (including National Health Service) services, resources, and support. Another bespoke questionnaire will ask participants to report on the actual academic marks and their academic targets for the previous year and whether they were satisfied with their academic progress. The Brief Resilience Scale is a brief and reliable means to assess the ability to bounce back from stress [47]. The Perceived Stress Scale-4 is a four-item measure of individuals perceived levels of stress and ability to cope [48]. The Adverse Events Questionnaire is a brief measure designed to assess stressful events in young people, which is proven to predict subsequent depression [49]. It consists of 3 questions asking about relevant adverse experiences (bad experiences concerning academic study; bad experiences concerning relationships; other bad experiences) rated from 0 “No”, 1 “yes, happened once”, 2 “yes happened twice”, 3 “yes, happened more than twice”. A fourth item asks about minor problems or hassles ranging from 0 “minor problems” to 4 “large number of minor problems and hassles”.
Participants will be asked to complete brief measures each week for eight weeks after randomisation to provide potential mediators of change: a two-item measure of depression (PHQ-2) [50]; a two-item measure of anxiety (GAD-2) [51]; a single item measure of stress over the last week (scored 0 none to 4 very severe); six items from the Cognitive and Behavioural Response to Stress Scale [52] assessing the frequency and usefulness of use of cognitive reinterpretation, behavioural activation, and relaxation or meditation strategies; 3-item adaptations of the automaticity component of the Self-Report Habit Index [53] focused on assessing the extent to which worry and problem-solving respectively are each a habit (e.g., “something that I do automatically" “I do without thinking”) over the past seven days; 2-items from the Self-Compassion Scale-short-form [54]; 2 items from the RRS-Brooding 5-item questionnaire [40]; a one-item measure assessing self-efficacy (“In the last seven days I feel better prepared to handle situations I could not handle before” rated using a seven‐point Likert scale ranging from −3 (“absolutely not true), 0 (“neither nor”) to +3 (“absolutely true”) and a one-item measuring problem clarification (“In the last seven days, I understand myself and my problems better” rated using a seven‐point Likert scale ranging from −3 (“absolutely not true), 0 (“neither nor”) to +3 (“absolutely true”).
The following descriptive variables will be assessed only at baseline: age, gender, sexuality, year of study, course of study, family’s occupational status, country of birth.
Sample sizeThe sample size was calculated using the primary outcome based on a minimum clinically important difference (MCID) at primary endpoint allowing for power at 0.80 and a two sided alpha at 0.05. The primary outcome is incidence of major depression across the 12 months follow-up. Assuming that the MCID is an absolute risk reduction in incidence of major depressive episodes of 10% based on previously published consensus [55] and that incidence of major depressive episodes will be 25% in the usual practice arm, paralleling previous studies in this high-risk sample [18, 19], and assuming 20% follow-up attrition, then conservatively we require n=324 per arm, giving a total sample required of 648.
Statistical analysis planThe primary analyses will be intention-to-treat (ITT) analyses [56] (i.e. all participants will be included in the analyses according to their randomised allocation) and based on complete case outcome data. The primary inferential analyses will compare across trial arms for the primary and continuous secondary outcomes at 12-months follow up using multilevel mixed-effect models, which enable us to examine nested hierarchies in the data (individual, intervention, university), across time (examining pre-to-post change), to capture dependencies in the data, to investigate individual trajectories (random intercepts, random slopes), and which have less restrictive assumptions re missing data.
Secondary analysis will be undertaken using both Complier Average Causal Effect models and multiple imputation:
Complier Average Causal Effect (CACE) analysis [57, 58] to provide an estimate of a treatment effect accounting for pre-specified per protocol adherence and compliance with the treatment, whilst retaining the benefits of randomisation.
Multiple imputation following the examination of the pattern of missing outcomes to impute primary and secondary continuous outcomes. Imputation models will be informed by treatment arm, baseline scores, other covariates to be included in the model, and other baseline characteristics found to predict outcome or propensity for missingness (logistic regression models will be used to investigate the associations between baseline characteristics and missingness).
Secondary analyses will be undertaken including:
Models will be estimated for the primary and secondary outcomes at 3- and 12-month follow-up and will include the covariates adjusted for in the linear regression models.
Cox regression models will be undertaken to investigate between group differences in the time until a major depressive episode.
Mediation analyses will be undertaken to gain insight into mechanisms that could explain the potential effect of the interventions on primary outcomes. We will use modern causal inference methods using structural equation modelling or parametric regression models to assess mediation effects [59]. In addition, we will investigate potential moderation of the interventions by site and history of depression.
The results of all models will be compared to primary analysis complete case ITT results.
Analyses will be undertaken by a statistician blinded to group allocation and using Stata v.17.
Organization, quality assurance and data managementResearch data will be automatically collected in a pseudonymised manner through an electronic data capture system licenced/programmed by Exeter Clinical Trials Unit (CTU). In the first instance all participants will be directed to the electronic data capture system to provide their data. Participants who respond to follow-ups by telephone will have their primary outcome data entered into the system by a site researcher and this variation in data collection method will be recorded in the EDC. All data will be kept securely and confidentially and only accessed by specified researchers at the University of Exeter. The central data-management team will use de-identified backups for the monitoring of the overall progress and data quality. Ultimately, a comprehensive de-identified dataset will be produced that includes all outcome data.
Trial statusThe Trial was registered in ISRCTN, number of identification 86795807. Date of registration: 27 October 2022. Recruitment commenced in July 2023 and is ongoing.
留言 (0)