
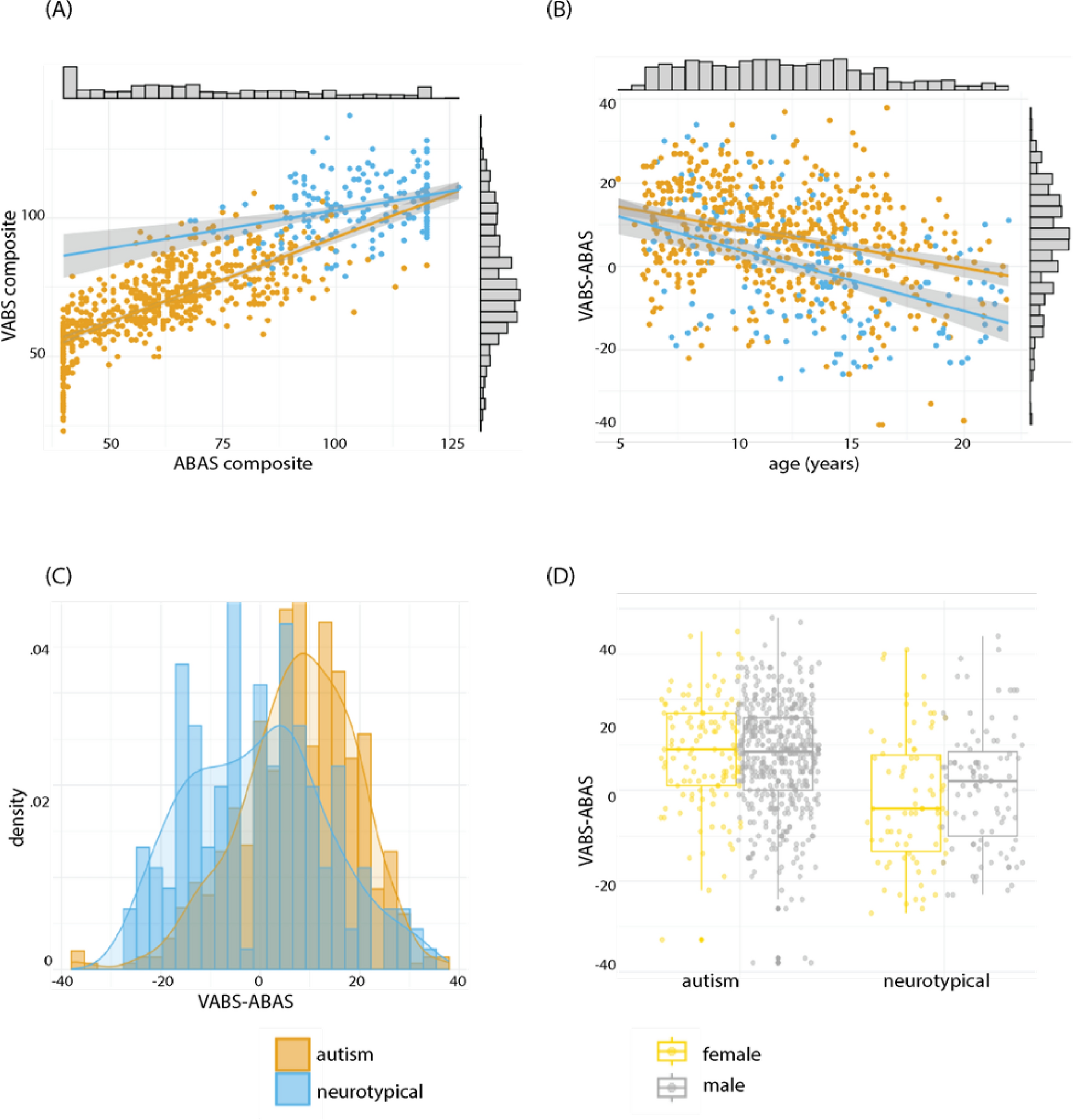
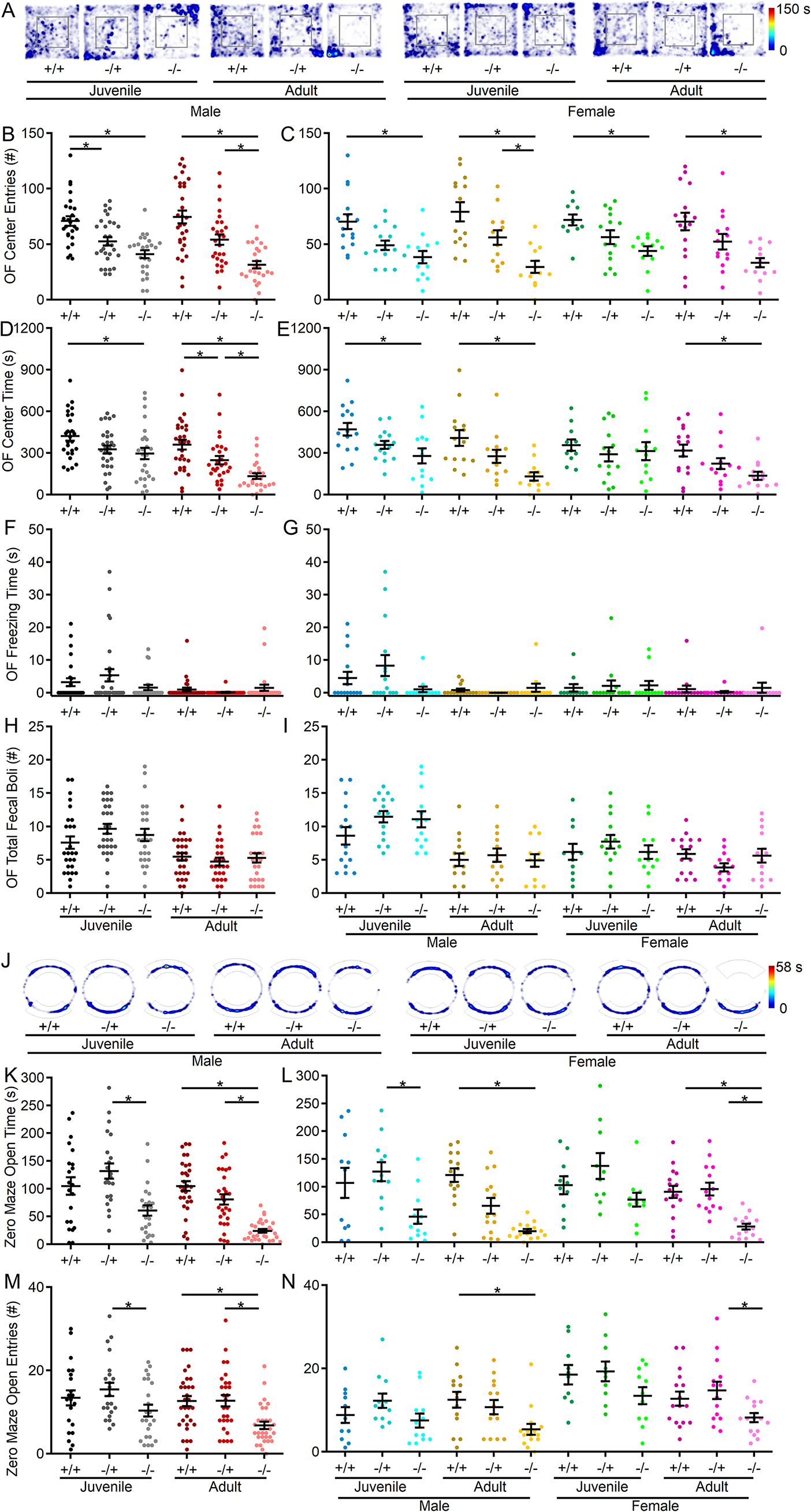
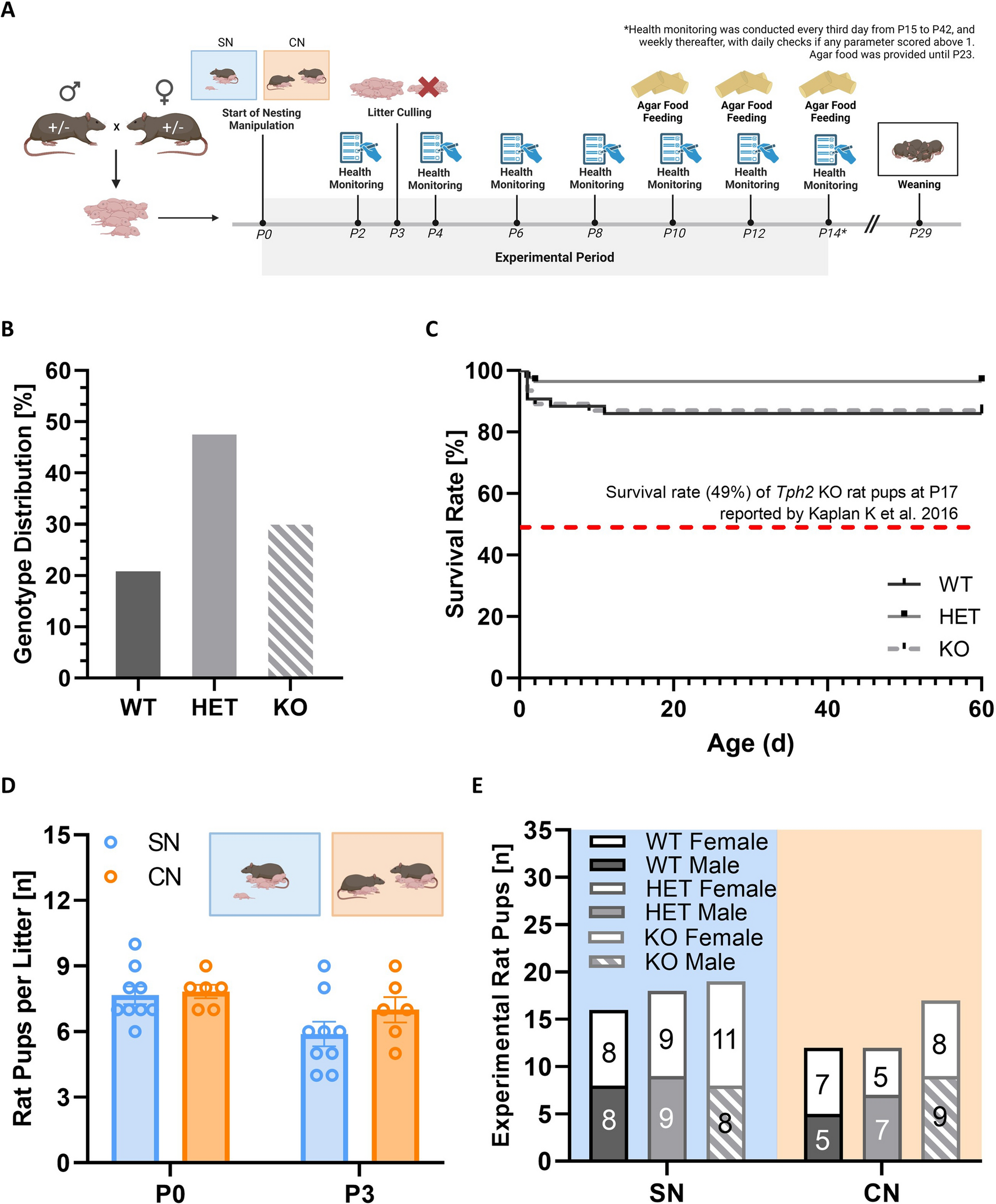
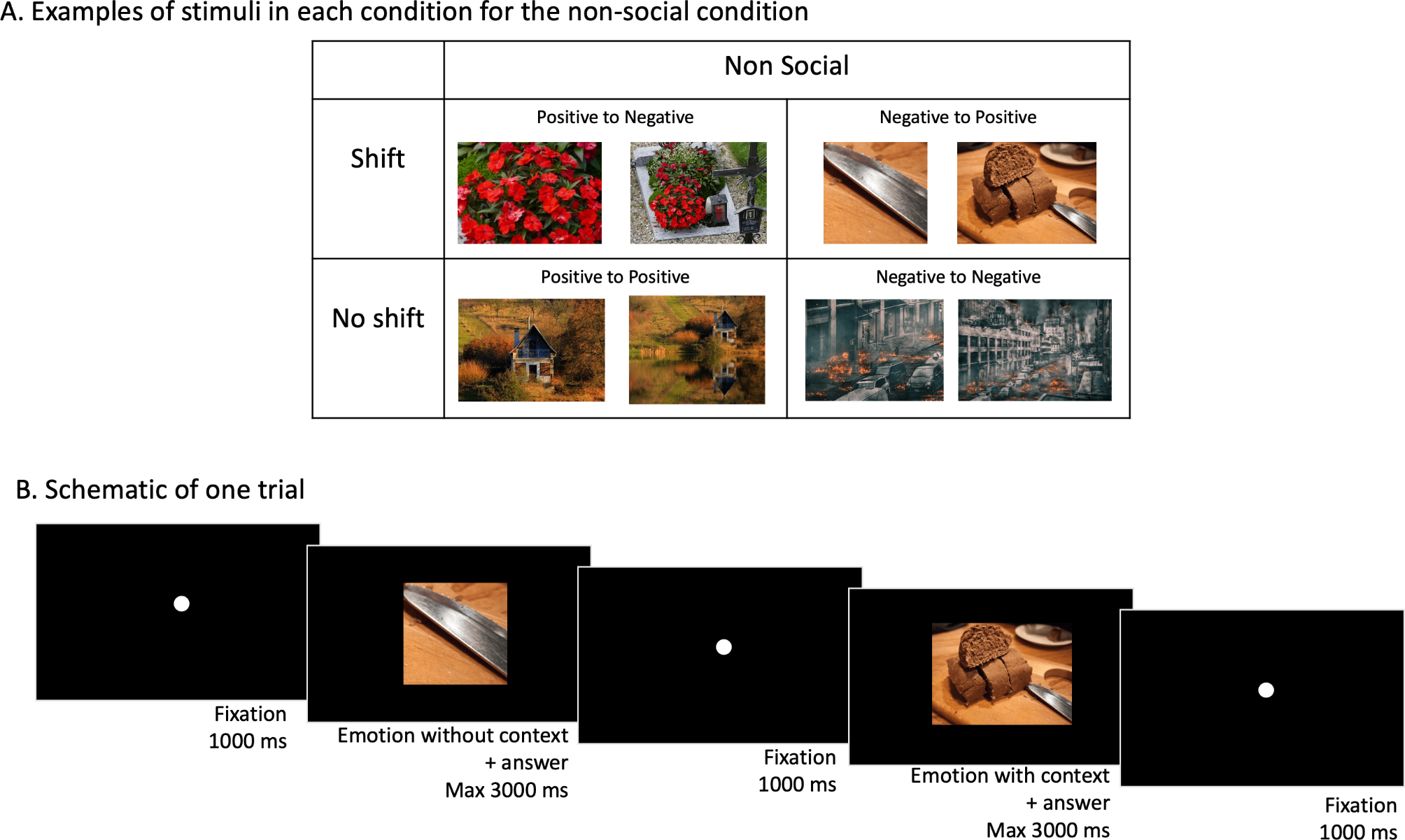
記住我






Independent CRISPR/Cas9 HDR transfections introduced the three SETBP1 SNVs into the KOLF2 iPSCs (Fig. 1A). HDR efficiency varied across the PATH2 SNV (3.86 ± 2.98%; n = 6), PATH3 SNV (9.32 ± 5.49%; n = 8) and VUS2 SNV (2.28 ± 1.64%; n = 8) transfections (Fig. 1B). Single cell clones were derived from the transfections with maximal HDR efficiency for PATH2 (8.37%) and PATH3 (16.36%); and VUS2 (4.31%, 4.54% and 3.21%). Derived cell clones were assessed with SETBP1 amplicon sequencing on genomic DNA (minimum of 30,000 reads), and analysed with CRISPResso2 software [11]. Three heterozygous (HDR/WT) clones for the PATH2 SNV and VUS2 SNV, and two heterozygous clones for the PATH3 SNV were generated (Fig. 1C). A further, six homozygous wild-type (WT/WT) clones were selected as isogenic controls.
Fig. 1
Generation of iPSC clones harbouring SETBP1 variants using CRISPR/Cas9 gene editing. A Schematic representation of the SETBP1 protein indicating the PATH2, PATH3 and VUS2 variants introduced into the KOLF2 iPSC genome using CRISPR/Cas9 gene editing. Five exons encode isoform A (1596 amino acid residues) of the SETBP1 protein. Truncating variants PATH2 and PATH3 are not located in any functional domains, however the missense variant, VUS2, is in an NLS region within the SET-binding domain. PEST: proline, glutamic acid, serine, and threonine rich sequence; NLS: nuclear localisation signal; Ath: AT hook; SKI: SKI homology region; HCF: HCF-1 binding motif; SET: SET-binding domain; Rpt: repeat. B CRISPR/Cas9 HDR efficiency for SETBP1 SNV integration into iPSC genomes. Data presented as mean ± s.d. HDR: homology directed repair; SNV: single nucleotide variant. C SETBP1 SNV and WT derived cell clones. Bar graphs show genotype for three heterozygous PATH2 variant cell lines, two heterozygous PATH3 variant cell lines, three heterozygous VUS2 variant cell lines, and the six wild-type clones selected as isogenic controls. SNV: single nucleotide variant; WT: wild-type; NHEJ: non-homologous end-joining. D Table of variant calling metrics for SETBP1 in NPC samples. Quality: Phred-scaled quality score
To confirm the presence of the variants, we applied the standard Genome Analysis Toolkit (GATK) best practices pipeline to call variants in our RNAseq data [16]. We were able to reliably detect all variants in our differentiated neural progenitor cells with high confidence (Phred-scaled probability that the site has no variant less than 10−33 for all variants). Data indicates the expression of both the normal wild-type and expected genetic variant transcript in each cell line (Fig. 1D, and Supplementary Fig. 1).
Next, clones were assessed at the genomic DNA level for chromosomal abnormalities by qPCR and were similar to parental KOLF2 iPSCs (Supplementary Fig. 2). Next, we examined the top six off-target sites for each SETBP1 crRNA using Sanger Sequencing (Supplementary Figs. 3, 4, 5) and determined normal gDNA integrity. All iPSC clones maintained a stem cell-like morphology during cloning (Supplementary Fig. 6), and the expression of stem cell markers OCT3 and NANOG (Supplementary Fig. 7).
SETBP1 PATH2, PATH3 and VUS2 genetic variants alter neural cell differentiationNeural differentiation was induced in each SETBP1 iPSC lines and cells harvested for analysis (Fig. 2A). Cell populations were analysed for pluripotency and neural marker expression after gating on cells, single cells, and then live cells for each timepoint across neural differentiation (Supplementary Fig. 7). Temporal changes in the percentage frequency expression of OCT3, PAX6 and NESTIN expression were determined (Fig. 2B). The proportion of cells expressing OCT3 was highest at day 0 of differentiation for SETBP1 PATH2 (82.03 ± 16.88%; n = 3), PATH3 (74.0 ± 2.43%; n = 3), VUS2 (87.33 ± 6.62%; n = 4) and WT (79.23 ± 5.40%; n = 6) populations. The frequency of OCT3 expression decreased across neural differentiation and there were no significant differences in the proportion of cells expressing OCT3, PAX6, or NESTIN at the iPSC (day 0) or NPC (day 24) stage of differentiation. Interestingly, at day 6 of neural differentiation, the percentage frequency of OCT3+ cells in SETBP1 PATH2 was decreased compared to WT (p = 0.005). The percentage frequency of PAX6 expression peaked at day 12 for SETBP1 PATH2 (18.41 ± 11.35%; n = 3), PATH3 (50.27 ± 20.42%; n = 3), VUS2 (62.13 ± 16.26%; n = 4) and WT (49.40 ± 17.38; n = 6) cells. Notably, there were significantly decreased PAX6 percentage frequency expression in SETBP1 PATH2 cells at day 12 (p = 0.008) and day 18 (p = 0.004) compared to the WT. NESTIN expression peaked at day 18 for SETBP1 PATH2 (81.37 ± 9.71%; n = 3) and at day 24 for SETBP1 PATH3 (84.4 ± 9.86%; n = 3), VUS2 (86.05 ± 6.30%; n = 4) and WT (86.42 ± 3.63%; n = 6) cells. There was an increase in the percentage frequency of NESTIN at day 12 in SETBP1 PATH2 cells compared to the WT (p < 0.001), and at day 18 in SETBP1 VUS2 cells compared to WT (p = 0.044).
Fig. 2
Differentiation of SETBP1 iPSC clones into neural progenitor cells. A Neural differentiation schematic showing differentiation of iPSCs into neural progenitor cells (NPCs) using neural induction media (NIM) and SMADi followed by neural progenitor media (NPM). Cells were harvested at selected timepoints across differentiation for pluripotency and neural marker analysis using flow cytometry and RNAseq. Image generated using BioRender. B Pluripotency and neural marker expression across neural differentiation. Graph plots indicate OCT3, PAX6 and NESTIN expression was assessed by flow cytometry in differentiating iPSC clones at 6-day intervals. Data presented as mean ± s.d.*p < 0.05; **p < 0.01; ***p < 0.001. WT: wild-type. C Morphology of NPCs derived from SETBP1 gene-edited iPSC clones. Representative images of NPCs harbouring PATH2, PATH3, VUS2 SETBP1 variants and WT NPCS at 4X objective magnification. D Representative images of neural marker, NESTIN and DCX, immunostaining in NPCs harbouring SETBP1 variants. Scale bar 50 µm. E Representative images of SETBP1 staining (green) relative to nuclear staining (blue) in iPSC-derived NPCs. Scale bar 50 µm
Throughout the neural differentiation there was a transition of cellular morphology observed, with large clustered iPSCs at day 0; to small, evenly dispersed, and elongated cells, with evidence of neurites projecting from the cell body, indicative of neural cells at day 24 (Fig. 2C). Next, using immunofluorescence staining we examined neural cell marker and SETBP1 expression. The neural markers NESTIN and DCX were expressed in both WT and SETBP1 genetic variant NPCs, with similar expression in WT, PATH2 and VUS2 cells, with increase expression in the PATH3 cells (Fig. 2D). Finally, in the NPCs, a nuclear pattern of SETBP1 expression was determined using immunofluorescence staining and this was consistent across the different SETBP1 genotypes (Fig. 2D, and Supplementary Fig. 8). Of note visually there appears to be reduced SETBP1 nuclear staining, that is more diffuse, in the SETPB1 PATH2 and SETBP1 PATH3 cells, when compared to the control or VUS2 cells. The SETBP1 PATH2 and PATH3 truncated proteins are likely subject to non-sense mediated decay, or as they may not localise to the nucleus efficiently due to the loss of two carboxy terminal NLS. Alternatively, the SETBP1 VUS2 is a missense mutation that may be stably expressed, and it retains all three NLS signals.
These data indicate temporal differences in neural progenitor cell differentiation from iPSCs harbouring SETBP1 PATH2 and SETBP1 VUS2 variants compared to the healthy matched control cells.
Transcriptomic data supported neural identity of differentiated iPSCsNext, we performed transcriptomics analysis to determine changes in gene expression and molecular and cellular pathways during neural cell differentiation. Principal component analysis (PCA) indicated clear separation between iPSCs and NPCs (Fig. 3A). To confirm derivation of NPCs from SETBP1 genetic variant and WT iPSCs, we integrated our transcriptomic data with publicly available data in the ARCHS4 database, and demonstrate that the SETBP1 WT, SETBP1 PATH2, SETBP1 PATH3, and SETBP1 VUS NPCs grouped with wild-type NPCs using PCA (Fig. 3B). Both experimentally derived and ARCHS4 NPCs align closely along principal components one and two, indicating a high degree of similarity.
Fig. 3
Neural identity of differentiated SETBP1 iPSC clones. A PCA plot demonstrating clear separation between iPSC and NPC samples based on gene expression. B PCA plot comparing SETBP1 experimentally derived NPCs with wild-type NPCs from the ARCHS4 database. C Numbers of DEGs up and downregulated in NPCs defined by SETBP1 genotype. D Analysis of top 50 significant upregulated genes in NPCs compared to iPSCs using Enrichr demonstrated enrichment of neural tissue gene sets in the ARCHS4 tissue database
There were 6101 differentially expressed genes (DEGs) between iPSCs and NPCs for SETBP1 WT cells, 5261 DEGS for SETBP1 PATH2 cells, 4872 DEGS for SETBP1 PATH3 cells, and 6201 DEGS for VUS2 cells, including up and down regulated genes (Fig. 3C; Supplementary Tables 4, 5, 6). Notably, at the transcript level there was down regulation of OCT3 and NANOG, and upregulation of neural transcripts including NES, MAP2, DCX, POU3F3, NR2F1, NR2F2, and SOX5 (Supplementary Fig. 9).
Analysis using EnrichR software and the top 50 upregulated genes during neural differentiation demonstrated enrichment of gene sets associated with neural tissues (Fig. 3D). The top four ARCHS4_tissues gene sets enriched during neural differentiation for SETBP1 WT cells were spinal cord (p = 1.75 × 10−11), spinal cord-bulk (p = 1.75 × 10−11), cerebellum (p = 8.28 × 10−10) and oligodendrocytes (p = 2.07 × 10−7). For SETBP1 PATH2 cells the top enriched gene sets were spinal cord (p = 1.55 × 10−10), spinal cord-bulk (p = 1.55 × 10−10), motor neuron (p = 7.07 × 10−7) and omentum (p = 7.07 × 10−7) while for SETBP1 PATH3 differentiation they were spinal cord (p = 9.67 × 10−9), spinal cord-bulk (p = 9.67 × 10−9), cerebellum (p = 3.35 × 10−8) and oligodendrocyte (p = 3.35 × 10−8). The top gene sets enriched for SETBP1 VUS2 cells across neural differentiation were spinal cord (p = 1.78 × 10−11), spinal cord-bulk (p = 1.78 × 10−11), motor neuron (p = 4.38 × 10−8) and cerebellum (p = 2.11 × 10−7). These data confirmed the differentiation of iPSCs into neural cells for each SETBP1 genotype consistent with findings observed for neural cell marker expression and cell morphology.
Disease pathways in SETBP1 genetic variant NPCsTo determine whether the SETBP1 genetic variant NPCs displayed a SETBP1-HD-like disease phenotype, we performed gene set enrichment analysis (GSEA) and identified DisGeNET and Disease Ontology (DO) gene sets enriched in SETBP1 variant NPCs compared to WT NPCs.
In DisGeNET, 67 gene sets were significantly dysregulated in SETBP1 PATH2 NPCs in comparison to WT NPCs (Supplementary Table 7). Gene sets related to nervous system disorders were significantly altered in genetic variant NPCs compared to WT NPCs (Fig. 4A) and included Hereditary Motor and Sensory Neuropathy Type 1; Charcot-Marie-Tooth Disease, Type Ia; and Holoprosencephaly, amongst others. In SETBP1 PATH3 NPCs compared to WT NPCs 655 gene sets were significantly dysregulated (Supplementary Table 8) including: Hereditary Motor and Sensory Neuropathy Type 1; Familial Dystonia; widened subarachnoid space; minicore myopathy with external ophthalmoplegia (disorder); and Generalized Hyperkinesia. In the SETBP1 VUS2 NPCs compared to WT NPCs, 57 gene sets were significantly dysregulated in the DisGeNET database (Supplementary Table 9). Similar to the SETBP1 PATH2 and PATH3 NPCs the VUS2 NPCs significantly dysregulated gene sets relating to nervous system disorders including Neuropathy; Holoprosencephaly; Orbital separation diminished; and mechanical allodynia. Other significantly dysregulated pathways in VUS2 NPCs included: Status Epilepticus; Neural Tube Defects; Midnasal Stenosis; and choanal atresia.
Fig. 4
Disease associated gene sets that are significantly dysregulated in SETBP1 variant NPCs compared to WT NPCs. Selected gene set terms that are significantly dysregulated in SETBP1 PATH2, SETBP1 PATH3 and SETBP1 VUS2 NPCs for DisGeNET (A) and Disease Ontology (B) databases. Full list of significant terms for each SETBP1 SNV is available in the supplementary data
In addition, we performed analysis using disease ontology (DO; Fig. 4B) database. The SETBP1 PATH2 NPCs had 23 gene sets significantly dysregulated compared to WT NPCs (Supplementary Table 10) and included sciatic neuropathy; motor neuron disease; holoprosencephaly; and peripheral nervous system disorder. In comparison to WT NPCs, SETBP1 PATH3 NPCs had a total of 367 DO gene sets dysregulated (Supplementary Table 11) including mononeuritis of lower limb; early infantile epileptic encephalopathy; muscular disease; and status epilepticus. The SETBP1 VUS2 NPCs had a total of 29 DO gene sets that were significantly dysregulated in comparison to WT NPCS (Supplementary Table 12). In SETBP1 VUS2 NPCs, DO pathway terms in common with SETBP1 PATH2 and SETBP1 PATH3 NPCs included lesion of sciatic nerve; mononeuritis of lower limb; sciatic neuropathy; and peripheral nervous system disease. DO terms unique to SETBP1 VUS2 NPCs included frontotemporal dementia; and substance related disorder.
This data identified many terms indicative of a SETBP1-HD-like phenotype. Holoprosencephaly is caused by incomplete separation of the prosencephalon (embryonic forebrain) to divide into double lobes of the cerebral hemispheres. Widened subarachnoid space is associated with motor and language delay. Other neural symptoms consistent with SETBP1-HD phenotype include hyperkinesia in ADHD; sensory changes in autism; dysmorphic facial features, ophthalmological abnormalities such as retinopathy; and alterations in digestion related pathways. Notably, a number of dysregulated pathways identified in the pathogenic truncation mutations, were also identified for the novel single nucleotide variant SETBP1 VUS2. However, there was a higher degree of similarity in the direction of enrichment score between SETBP1 VUS2 and SETBP1 PATH2 NPCs compared to SETBP1 PATH3 NPCs.
Variants in SETBP1 cause a specific perturbation to the transcriptomeTo further investigate changes in cellular pathways we examined differential gene expression (DEG) and performed GSEA. SETBP1 PATH2, SETBP1 PATH3 and SETBP1 VUS2 NPCs were compared to WT controls to determine significant pathway gene set changes in Gene Ontology (GO)-Biological Process (Fig. 5A), GO Cellular Component (Fig. 5B), and GO Molecular Function (Fig. 5C).
Fig. 5
Gene sets using Gene Ontology terms that are significantly dysregulated in SETBP1 variant NPCs compared to WT NPCs. Selected gene set terms that are significantly dysregulated in SETBP1 PATH2, SETBP1 PATH3 and SETBP1 VUS2 NPCs for Gene Ontology (GO)-Biological Process (A), GO-Cellular Component (B), and (C) GO-Molecular Function terms. GSEA plots demonstrate opposing patterns of gene set expression between PATH2 and VUS2 compared to PATH3 for GO-BP terms forebrain development (D) and regulation of neural precursor cell proliferation (E). Likewise, gene set expression for GO-MF term frizzled binding (F) was aligned for PATH2 and VUS2 and contrasted with PATH3. Full list of significant terms for each SETBP1 SNV is available in the supplementary data
In initial gene expression analysis, the SETBP1 PATH2 variant had 642 DEGs in comparison to WT NPCs, including 332 genes that were upregulated and 310 genes that were downregulated (Supplementary Table 13). In contrast, SETBP1 PATH3 NPCs had no DEGs compared to WT, while a single gene was significantly differentially expressed in SETBP1 VUS2 NPCs compared to WT NPCs: TRIM4 (logFC: − 3.86; p = 6.23 × 10−7) in comparison to WT NPCs. Next, as there were temporal changes in neural markers during NPC differentiation, we determined the DEG for the difference of differentiation, comparing the changes in SETBP1 PATH2 differentiation to SETBP1 WT differentiation. There were 32 significant DEGs (Supplementary Table 13), and the top upregulated gene was ISL2 a negative regulator of neuron differentiation.
In GO Biological Process (GO-BP) terms, comparison of genetic variant SETBP1-like NPCs to WT NPCs demonstrated significant pathway dysregulation (Supplementary Tables 14, 15, 16) in neural networks associated with forebrain development; neural cell specification; and nervous system development. GO-BP significant gene sets common to SETBP1 PATH2, PATH3 and VUS2 NPCs in comparison to WT cells included: pattern specification process; forebrain regionalization; cell proliferation in forebrain; trigeminal nerve development; forebrain development; dopaminergic neuron differentiation; regulation of neural precursor cell proliferation; telencephalon development; spinal cord dorsal/ventral patterning; diencephalon development; and central nervous system neuron differentiation. Interestingly, the direction of enrichment scores in forebrain development (Fig. 5D) and regulation of neural precursor cell proliferation (Fig. 5E) for each SETBP1 genotype indicate similarity between SETPB1 PATH2 and SETBP1 VUS2 NPCs.
In further analysis of GO-BP gene pathways, we investigated genes from all common pathways between SETBP1 variants using STRING and identified a central link between SETBP1 pathways genes and GATA2 (Fig. 6). The importance of GATA transcription factors and ability to replace OCT4 to induce pluripotency in somatic cells has been previously reported [17], whilst other research indicates GATA2 has a role in neural specification [18]. Interestingly, in the DEG comparison of SETBP1 PATH2 NPC to iPSC, GATA2 was among the downregulated DEG (logFC = − 3.4782, adjusted p value = 0.0054; Supplementary Table 4).
Fig. 6
STRING network demonstrating protein associations in dysregulated GO-BP gene sets across the SETBP1 variant NPCs. GATA2 was identified as a key transcription factor linking to SETBP1 and many of the core enrichment genes in dysregulated gene sets in SETBP1 variant NPCs. Proteins directly associated with GATA2 are shown in orange, while proteins indirectly associated with GATA2 are shown in yellow. Level of confidence in association between proteins is represented by the thickness of the line
In GSEA GO-Cellular Component (GO-CC; Fig. 5B), gene sets that were significant for SETBP1 PATH2 NPCs compared to WT (Supplementary Table 17) included neuronal cell body; axoneme; and apical junction complex. In SETBP1 PATH3, significant GO-CC terms (Supplementary Table 18) included apical junction complex; ion channel complex; and transporter complex. In SETBP1 VUS2 NPCs compared to WT, significant pathways were overlapping with SETBP1 PATH2 and SETBP1 PATH3 terms including apical junction complex; neuronal cell body; cell–cell junction; apicolateral plasma membrane; and tight junction. Specific to VUS2 were the gene sets distal axon; neuronal dense core vesicle; and neuron projection terminus (Supplementary Table 19).
The ion channel complex and transporter complex are responsible for selective cell permeability and active transport. Recent findings indicate that these systems co-operate in cell ion homeostasis and can physically interact with neurotransmitter transporters expressed in the brain [19] to form a cellular signalling hub.
In GO Molecular Function (Fig. 5C), gene sets that were significant for SETBP1 PATH2 NPCs compared to WT (Supplementary Table 20) included structural constituent of ribosome; signalling receptor regulator activity; and frizzled binding. The significantly dysregulated gene sets for PATH3 NPCs (Supplementary Table 21) included: growth factor binding; integrin binding; and extracellular matrix structural constituent. In VUS2 NPCs (Supplementary Table 22) pathway gene sets overlapped with PATH2 NPCs including frizzled binding; DNA-binding transcription activator activity, RNA-polymerase II-specific; and signalling receptor regulator activity. The enrichment scores for frizzled binding indicate similarity between PATH2 and VUS2 NPCs (Fig. 5F).
The Frizzled (FZD) pathway is stimulated by WNT ligand binding to the receptor leading to canonical and noncanonical Wnt signalling [23]. Ligands represented in the core enrichment gene sets for both PATH2 and VUS2 NPCs included WNT2B, WNT3/3A, WNT4, WNT7A/B, WNT8A/B and WNT9A/B (Suppl Tables 20 and 22). These factors have a variety of roles in brain development, including patterning and regionalisation [20], neural stem cell self-renewal and differentiation [21], neuronal migration [20], axon guidance and synapse formation [22]. Accordingly, dysfunction of WNT signalling due to SETBP1 mutation likely perturbs neural cell development and function. Components of both the Wnt and the related Hippo Signalling pathways are affected in a SETBP1 variant-dependent manner (Supplementary Fig. 10). In other studies, SETBP1 is identified as a part of a DNA-binding complex that modulates histone methylation to make chromatin more accessible and regulate gene expression. Recent studies propose an epigenetic complex consisting of POLR2A, PHF8/6, MLL1, HCF and SETBP1 that regulates transcription according to histone methylation status at H4K20me and H3K4me [23].
These data indicate that the SETBP1 PATH2, SETBP1 PATH3 and SETBP1 VUS2 NPCs have common GO terms, potentially highlighting overlapping mechanisms of cell dysfunction that may contribute to SETBP1-HD.
留言 (0)