
記住我







We first re-analyzed the stromal subset (12,584 cells) from the 1st trimester selective termination scRNA-seq data from [1]. We identified five clusters instead of the three previously described clusters [1] (Fig. 1A). Our clustering analysis separated the original dS1 cluster into two distinct clusters, namely dS1A and dS1B, where dS1A consisted of the most fibroblastic type of stromal cells expressing ID2 and IGF1 (Supplementary Table S1). The decidualizing dS2 cluster expressed many classical pre-decidual markers such as LEFTY2 and FOXO1 (Fig. 1B, Table S1). Furthermore, the expression of prolactin (PRL) and IL1B confirmed the identity of the decidualized dS3 (DSC) cluster (Fig. 1B). However, recent studies in vitro have shown branching differentiation trajectories of decidual cells, resulting in a decidualized population and a senescent decidual population [5, 18], but this senescent population was not detected in the previous 1st trimester pregnancy data analysis. Here we identified a cluster with upregulation of senescence markers such as IL32 and CXCL8 (Fig. 1B, Supplementary Table S1), and apoptosis markers such as G0S2 (Supplementary Fig. 1), which were previously detected in assembloids in vitro [18]. This cluster also expressed several NK cell markers, including GNLY and NKG7. This suggests that these cells either express NK markers or alternatively these trancripts could originate from NK cells. Therefore, we annotated this cluster as senescence/NK associated decidual cells (dSsen/nk).
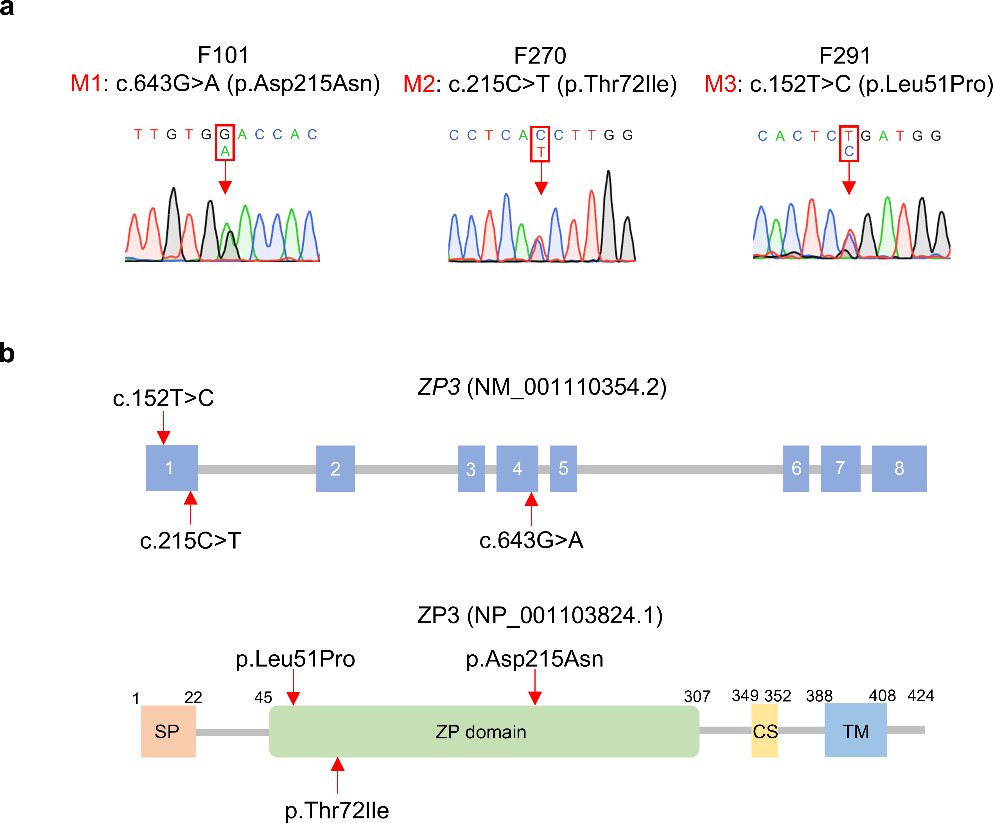
Fig. 1
Gene regulatory network analysis of 1st trimester pregnancy decidual stromal (dS) cells. A UMAP visualization of the cluster analysis of the stromal single-cell transcriptomes, data from [1] B Top five maker genes of the stromal cluster with FDR 0.05 and FC ranking. dS1A and dS1B represent the undifferentiated fibroblastic state, dS2 the decidualizing state, dS3 the decidualized state and dSsen/nk the senescent/natural killer cell interacting cells. C A heatmap for single-cell gene regulatory network analysis (SCENIC) result for the top 10 cluster specific regulons (transcription factor + target genes). The column indicates subpopulation specific cell clusters (mRNA expression based) based and the rows indicate the regulon TF with number of target genes (g). See Supplementary Table S2 for regulon specificity scores. The dot plot displays the cell cluster specific mRNA expression of the TFs. On the right the selected cluster specific regulon TFs are marked with a bar. D Hierarchical clustering of selected enriched gene ontology (GO) terms for combined predicted dS3 regulon (BRF2, DDIT3, ZNF274 and ZNF226) targets versus predicted dSsen/nk regulon (SMAD1, PPARA, MDM2, ETV6 and EOMES) targets. See Supplementary Table S3 for detailed regulon target gene lists
Cluster Specific Transcriptomic Regulators of Decidual Stromal CellsTo gain insight into the major transcriptional regulators relevant to the 1st trimester stromal subpopulations, we performed a gene regulatory network analysis using SCENIC. We then displayed the top 10 stromal subpopulation-specific regulons, consisting of a TF and its predicted target genes (Fig. 1C, Supplementary Table S2). Using a heatmap depicting the binary regulon activity score for each cell, we identified whether the TF was active or not in a given cell. Our results revealed that a considerable proportion of the top 10 subpopulation-specific regulons were partially shared among the subpopulations. However, we also detected subpopulation-specific regulons (Fig. 1C). The subpopulation-specific activity scores of the top regulons were generally consistent with the scRNA-seq data, such that expression of the regulon TF itself peaked in the corresponding subpopulation. Furthermore, we observed that for many of the detected regulons, both the active regulon score and TF expression were only present in a fraction of the cells within the cluster. This might be partly due to incomplete detection of moderately or lowly expressed genes in scRNA-seq data.
For the undifferentiated fibroblastic subpopulations (dS1A, dS1B), we detected regulons such as POLR2A and SFR for dS1A, and FOSL1, BHLH40, MAFF, KLF6, and KLF4 for dS1B. In functional enrichment analysis, the predicted target genes in these regulons were associated with undifferentiated proliferative functions like "blood vessel morphogenesis" (p = 1.99 × 10–9) and "regulation of cell junction assembly" (p = 2.67 × 10–9) for dS1A, and "hematopoietic or lymphoid organ development" (p = 3.76 × 10–22) for dS1B (Supplementary Table S3).
Regulon TFs and Networks for the Decidualizing Stromal Cells (dS2)Next, we investigated the decidualizing stromal cell population dS2 in more detail. Our regulon analysis revealed a cluster of seven core regulons, which included PRDM1 (Blimp1), FOXP1, NFE2L1, SOX5, STAT2, ARID3A, and PBX3 (Fig. 1C and Supplementary Fig. 2). Of these regulons, especially the transcription factors PRDM1, FOXP and NFE2L1 had dS2 specific expression (Fig. 1C and Supplementary Fig. 2). Moreover, when the regulon activity on/off score and the expression of the TF itself was visualized with UMAPs, the regulon activity score of PRDM1, FOXP1 and NFE2L1 regulons matched more accurate than the TF expression with the cells in the dS2 subpopulation cluster (Fig. 1A and Supplementary Fig. 2). PRDM1, a known decidualization TF in mice [21], had several predicted target genes involved in decidualization, such as HAND2, LEFTY2, WNT5A, PRLR, and IGFBP2 (Supplementary Fig. 2, Supplementary Table S2). FOXP1 also has predicted targets LEFTY2 and PRLR, and additionally IL15, which has a crucial role in the stromal induced maturation of dNK cells [10]. NFE2L1 (NRF2), together with PRMD1, have as predicted targets decidualization genes such as follistatin (FST) [22], insulin receptors substrate 2 (IRS2) [23], and GADD45G (DDIT2). The functional enrichment analysis of the combined dS2 core regulons identified significant enrichment of GO terms such as “response to hormone” (p = 1.02 × 10–22) and “regulation of Wnt signaling pathway” (p = 3.01 × 10–20) (Supplementary Table S3).
Fig. 2
Gene regulatory networks of selected dS3 specific regulons. The size of the TF node is proportional to the number of target genes that the TF regulates, the target set is filtered to contain only cluster specific upregulated genes (FDR 0.05). The arrows indicate the target genes for the TF, and the different colors of target genes indicate target groups defined by the regulating TF or combinations of TFs. The UMAPs display the TF mRNA expression (left) and the binary regulon activity (on/off) (right) in each cell
Regulon TFs and Networks for the Decidualized Stromal Cells (dS3)For the decidualized stromal cell population dS3, we detected four core regulons, including DDIT3, BRF2, ZNF274, and ZNF226, of which the TF expression peaked in dS3 for DDIT3, BRF2, and ZNF226 (Fig. 1C). Notably, the cells where DDIT3 and BRF2 regulons were active showed the highest PRL expression (Fig. 2), confirming that their activity aligns accurately in vivo with cells that are decidualized, as indicated by the classical PRL decidualization marker. To our knowledge, these TFs have not previously been directly linked with decidualization, but their known functions align with the known decidual functions. DDIT3 is a known regulator of endoplasmic reticulum (ER) stress and unfolded protein stress responses [24] and a prolactin target gene in mouse decidua [25]. In our analysis, DDIT3 is predicted to target both decidual markers PRL, IL1R2, and DDIT4 (Fig. 2), as well as stress response proteins such as heat shock proteins HSPA5 (HSP70 family), and HSPB1 (HSP20 family) (Supplementary Table S3). BRF2 is a major redox-sensing transcription factor involved in oxidative stress protection [26]. Predicted BRF2 targets also included genes related to hormone-related stress responses such as HSD11B1, which is involved in the synthesis of the stress hormone cortisol. BRF2 and DDIT3 shared common predicted targets, such as MAOA, NDP, TM4SF1, and PRR15 (Fig. 2), of which monoamine oxidase MAOA is known to regulate the differentiation of endometrial epithelial cells [27], whereas NDP and TM4SF1 are part of wnt/beta-catenin signaling. BRF2 was also predicted to target secretory molecules, such as WNT4, typical to the secretory endometrium [28].
Senescence/NK Associated Stromal Cells (dSsen/nk) are Predicted to be Targeted by NK CellsFor dSsen/nk, we identified five core regulons, including EOMES, ETV6, MDM2, PPARA, and SMAD1, of which TF expression peaked in dSsen/nk for EOMES and MDM2. EOMES is a known TF in NK cell differentiation [29] and regulates NK-mediated granulysis. The detection of several NK cell markers in this cluster, such as GLNY and NKG7 (Fig. 1B, Supplementary Table S3), supported the interpretation that mRNAs in the cells of this stromal cluster could partly originate from NK cells. Furthermore, NK cells are known to transmit protein and mRNA [3, 30]. Thus, because of the possibility of mRNA import, and the small number of cells in this cluster, we did not further focus on the GRNs of this cluster but conducted a functional enrichment analysis of the predicted core regulon targets to generally identify the functional differences between dSsen/nk and dS3. GO terms significantly enriched in dS3 but not dSsen/nk predicted targets included “regulation of response to endoplasmic reticulum stress” (p = 7.67 × 10–8) and “regulation of steroid hormone secretion” (p = 5.54 × 10–7) (Fig 1D, Supplementary Table S3), reflecting a transcriptional state of decidualized dS3, where these cells have acquired higher resistance for stress, in line with previous reports [4]. On the other hand, the dSsen/nk specific terms included “cytolysis” (p = 2.67 × 10–10) and positive regulation of chemotaxis (p 3.84 × 10–7) (Fig 1D, Supplementary Table S3). The detected “cytolysis” functional term for dSsen/nk is supported by the dSsen/nk specific upregulation G0S2 (Supplementary Fig. 1), that was recently reported as apoptosis inducer in decidualizing stromal cells [31], overall supporting the prediction that the dSsen/nk subpopulation is targeted for apoptosis and immunoclearance by the dNK cells [3].
Reannotation of the dNK Clusters Using scRNA-seq DataNext, we conducted a re-analysis of the dNK subset (11,884 cells) from the 1st trimester selective termination scRNA-seq data from [1]. We identified six clusters (Fig. 3A). Our cluster assignments for dNK1, dNK2, dNK3, and NK proliferative (NKp) were consistent with those from the original study [1]. Therefore, we adopted the same nomenclature. Our analysis also revealed two additional smaller adjancent clusters, which we annotated based on expression markers and regulon analysis (Fig. 3B) as dNK MAF + (155 cells) and dNK NRF2F2 + (134) clusters. The NRF2F2 + cluster had strong expression of typical stromal markers, but it clustered together with other dNK cell clsuters. For dNK MAF + cells the strong MAF transcriptional signature, a major macrophage marker, suggests that dNK MAF + cells either express macrophage markers or alternatively these transcripts could originate from macrophages. For both of these smaller adjacent clusters we did not proceed to full SCENIC analysis and focused on the major dNK clusters with > 1000 cells that are predicted to be functionally most relevant.
Fig. 3
Gene regulatory network analysis of 1st trimester pregnancy decidual natural killer (dNK) cells. A UMAP visualization of the cluster analysis of the dNK single-cell transcriptomes [1] B Top five maker genes of the stromal cluster with FDR 0.05 and FC ranking. C A heatmap for single-cell gene regulatory network analysis (SCENIC) result for the top 10 cluster specific regulons (transcription factor + target genes). The column indicates subpopulation specific cell clusters (mRNA expression based) based and the rows indicate the regulon TF with number of target genes (g). See Supplementary Table S5 for regulon specificity scores. The dot plot displays the cell cluster specific mRNA expression of the TFs and on the right the selected cluster specific regulon TFs are marked with a bar. See Supplementary Table S6 for detailed regulons target gene lists
Cluster Specific Transcriptomic Regulators of dNK CellsIn order to understand the major transcriptional regulators specific to the 1st trimester dNK subpopulations, we ran SCENIC similarly as for the stromal cells and depicted the top 10 subpopulation-specific regulons using a heatmap (Fig. 3C). The dNKp population displayed a robust cell cycle regulator signature (E2F1, E2F2, E2F7 and E2F8). The regulon signatures for the other clusters were more subtle (Fig. 3C, Supplementary Table S5). Notably, in the dNK2 population, with the greatest number of cells, the vast majority of the regulons were shared with both dNK1 and dNK3. To emphasize the most relevant functional differences, we further investigated the divergent dNK1 and dNK3 subpopulations, with focus on the core subpopulation-specific dNK regulons where the mRNA expression of the regulon TF also peaked in the concordant subpopulation (Fig. 3C, 4 and 5).
Fig. 4
Gene regulatory networks and regulon activities of selected dNK1 specific regulons. The size of the TF node is proportional to the number of target genes that the TF regulates, and the target set is filtered to contain only dNK1 upregulated genes (FDR 0.05). The arrows indicate the target genes for the TF, and the different colors of target genes indicate target groups defined by the regulating TF or combinations of TFs. The UMAPs display the TF mRNA expression (left) and the binary regulon activity (on/off) (right) in each cell
Fig. 5
Gene regulatory networks and regulon activities of selected dNK3 specific or dNK2/dNK3 specific regulons. The size of the TF node is proportional to the number of target genes that the TF regulates, and the target set is filtered to contain only dNK3 upregulated genes (FDR 0.05). The arrows indicate the target genes for the TF, and the different colors of target genes indicate target groups defined by the regulating TF or combinations of TFs. The UMAPs display the TF mRNA expression (left) and the binary regulon activity (on/off) (right) in each cell
In functional enrichment analysis, we observed that the predicted regulon targets of dNK1 included genes for development for the uterus and immunotolerance, whereas the predicted dNK3 regulon targets were enriched with inflammation related GO terms (Supplementary Table S6). Specifically, for dNK1, these included “in utero embryonic development” (p = 2.45 × 10–9) and “response to hormone” (p = 1.47 × 10–7), whereas the dNK3 top terms were “response to virus” (p = 9.33 × 10–34) and “defense response to symbiont” (p = 9.05 × 10–26) (Supplementary Table S6).
Predicted dNK1 Regulon Targets Suggest that dNK1 Cells Promote Maternal ImmunotoleranceThe predicted dNK1 core regulons RELB, IRX3, FOXP2, and RREB1 and regulon targets that were up-regulated (FDR 0.05) in dNK1 cluster are displayed in Fig. 4. RELB is a negative regulator in NFKB pathway and is predicted to target other anti-inflammatory TFs such as STAT3. The RELB targets include TNFRSF18 receptor, CSF1, which promotes immunotolerance by interacting with extravillous trophoblasts [1, 32] and SPDL1, which reduces cytotoxicity of maternal immune cells [1, 33] (Supplementary Table S6).
IRX3 was detected as the top-ranked dNK1 specific regulator. It is predicted to target ENTPD1 (CD39), which has high dNK1 specific expression and was reported to convert extracellular ATP to adenosine [1, 34], thereby downregulating cytotoxic extracellular ATP buildup. In addition to direct immunomodulation, it has been reported that dNK1 cells are metabolically reprogrammed to induce glycolysis pathway [1]. IRX3 and RREB1 are predicted to target EPAS1 (HIF-2alpha), which is upregulated in dNK1, and HIFs are the known main glycolysis regulators. Moreover, predicted FOXP2 targets are enriched in GO terms such as “response to oxygen levels” (p = 1.38 × 10–4). Therefore, all three IRX3, RREB1 and FOXP2 may be involved in promoting metabolic reprogramming in dNK1.
Furthermore, RELB, RREB1 and FOXP2 all target CDKN1A, a senescence inducer, raising the possibility that dNK1 population or dNK cells, in general, not only survey stromal cells for senescence but also themselves are programmed for it.
Predicted dNK3 Regulon Targets Suggest that dNK3 Cells Promote InflammationThe predicted dNK3 core regulons TBX21 (T-bet), IRF2, TGIF1 and FOXN2, and predicted regulon targets that were upregulated (FDR 0.05) in dNK3 cluster are shown in Fig. 5. These regulons include TFs that are well-known regulators of NK development such as TBX21 (T-bet) and IRF2. TBX21 is predicted to target several genes related to “leukocyte migration” (p = 3.28 × 10–7) including CCR5, LCK, PIK3R1, SLAMF1, TBX21, CRTAM, PREX1 (Supplementary Table S6). TBX21 together with TGIF1 are predicted to target TAGAP, that is a known regulator of T cell differentiation and autoimmune diseases [35].
We observed that dNK2 and dNK3 shared multiple predicted regulons related to inflammatory signaling that promote NK cell cytotoxicity. These included transcription factors related to interferon signaling such as IRF1, IRF2, IRF5, IRF6, IRF6, IRF7 and IRF9 (Supplementary Table S5), with IRF7 being most notable for dNK2 and IRF2 for dNK3. The predicted targets of IRF2 and IRF7 included a wide range of genes involved in interferon response, such as IFNG and IFNA signaling mediators, cytokines such as CCL4, CCL5, CCL8, CCL19, XCL1, CXCL10, CXCR4, IL33, IL16 and CCL4L2, and core inflammatory regulators such as STAT1 and TNF (Fig. 5, Supplementary Table S6). IRF2 also regulated GADD45G, which has been reported to enhance IFNG production and anti-tumor immune effects [36].
In addition TGIF1 (TGFB Induced Factor Homeobox 1) was predicted to target genes involved in TGFB pathway (TGIF1, PPP1R15A, PMEPA1, GREM2) and genes in GO term “regulation of ERK1 and ERK2 cascade (p = 1.14 × 10–5) (Supplementary Table S6). Previously studies have shown that TGFB signaling is associated with the repression of NK-mediated cytotoxicity [37], suggesting the possibility of dNK3-specific TGFB-mediated immunotolerance mechanisms that are distinct from those indicated for dNK1.
Pregnancy Disorder Downregulated Genes are Enriched Among Predicted dS2 / dS3 Regulon Target Genes and Disorder Upregulated Genes Among Predicted dNK2 / dNK3 Regulon TargetsWe analyzed the translational relevance of the identified regulons by intersecting their predicted target genes with pregnancy disorder datasets. We utilized overrepresentation analysis to determine if the predicted regulon target genes were enriched among disorder upregulated or disorder downregulated genes (Fig. 6, Supplementary Table S7). The disorders analyzed included “severe preeclampsia, including early-onset (EOP, before week 34) and late-onset preeclampsia (LOP)”, “recurred pregnancy loss (RPL)”, or “missed abortion (MA)”. We found that for stromal cells, the predicted dS2 and dS3 specific regulon targets were enriched with preeclampsia downregulated genes, particularly regulon targets predicted for PRDM1 (LOP down p = 1.75 × 10–18, 21-fold) and for FOXP1 (LOP down, p = 3.8 × 10–14, 9.1-fold) (Fig. 6A, Supplementary Table S7). Figure 6B presents the mRNA expression of these predicted PRDM1 and FOXP1 targets in specific cell subpopulations. These include decidualization markers such as PRLR and IGFBP3, as well as genes that have previously been reported to have roles in trophoblast invasion, such as FERMT2 [38] and RORB, for which expression changes in preeclampsia have been observed in fetal placenta [39].
Fig. 6
Overrepresentation analysis reveals pregnancy disorder associated cell subtype -specific regulons. A Overrepresentation of stromal dS2 and dS3 regulon target genes in pregnancy disorder datasets. The values in the cells are overrepresentation p values from Fisher’s exact test with all Refseq protein-coding genes (n = 20,203) as the background. The arrows on the x-axis represent up or down regulated disease genes in the corresponding bulk transcriptomic data. These included Early-Onset severe Preeclampsia (EOP) and Late-Onset severe Preeclampsia (LOP) from [20], and a study that contrasted women with previous severe preeclampsia to controls with normal pregnancy that were posteriorly collected during late secretory phase of Menstrual cycle (PP-M) [13]. B dS cell subpopulation specific mRNA expression of predicted PRDM1 and FOXP1 targets that were overrepresented in LOP downregulated genes. The specific PRDM1 and FOXP1 target groups are indicated on the right. C Overrepresentation of dNK1, dNK3, dNK2/3 regulon target genes in pregnancy disorder datasets. The values in the cells are p values from Fisher’s exact test with all Refseq protein-coding genes as the background. The arrows on the x-axis represent up or down regulated disease genes in the corresponding bulk transcriptomic data. These additionally included missed abortion (recurrent pregnancy loss) MA(RPL) data from [9], and unexplained recurred pregnancy loss enriched /underrepresented cluster specific genes (URPL) from [12]. D dNK cell subpopulation specific mRNA expression of IRF2 and IRF7 target genes that were overrepresented in MA(RPL), EOP or LOP upregulated genes. The condition in which the genes were upregulated is displayed on the left. On the right, IRF2 and IRF7 target groups are marked together with the specific classical IFNA or IFNG target genes. For the gene lists used as inputs and results of Fisher’s tests see Supplementary Table S7
In dNKs, we observed that the predicted dNK2/3 regulon targets were enriched with disorder upregulated genes whereas the predicted dNK1 regulon targets were enriched with pregnancy disorder downregulated (/underrepresented) genes (Fig. 6C). For the pregnancy disorder upregulated regulons and target genes, we identified that the predicted dNK2/3-associated IRF2 and IRF7 targets were enriched with preeclampsia upregulated genes (IRF2 LOP p = 9.2 × 10–5, 19-fold; IRF7 EOP/LOP (p = 5.0 × 10–5/5.6 × 10–5), 7.1-fold / 8.6-fold) (Fig. 6C, Supplementary Table S7). Figure 6D illustrates the cell subpopulation-specific mRNA expression of these predicted targets, including CD2, ISG20, and CCL5, which are involved in proinflammatory responses. Notably, the cell surface receptor CD2, which mediates nanotube formation [40] and augments cytotoxicity [41], is a target of both IRF2 and IRF7 and was found to be upregulated in both preeclampsia and missed abortion. Furthermore, we found several predicted IRF7 targets enriched with missed abortion upregulated genes (p = 4.6 × 10–5, 3.7-fold) including classical interferon gamma and alpha response genes (CXCL10, GBP2, MX1, MX2, PARP9, RSAD2, SAMD9L) (Fig. 6D, Supplementary Table S7). These results support previous observations that upregulation dNK2/3 subpopulation specific transcriptomic states promote pregnancy disorders and emphasize the potential role of over-activated interferon pathway in the etiology of these disorders.
For the pregnancy disorder downregulated regulons and target genes, for dNK1, we found that that predicted FOXP2 targets were enriched with genes downregulated in missed abortion (RPL) [19], (p = 1.8 × 10–4, tenfold, LAYN, EGLN2, FOXP2, LAMA4 and SPARC) and predicted RREB1 targets were enriched with unexplained recurrent pregnancy loss underrepresented cluster [
留言 (0)