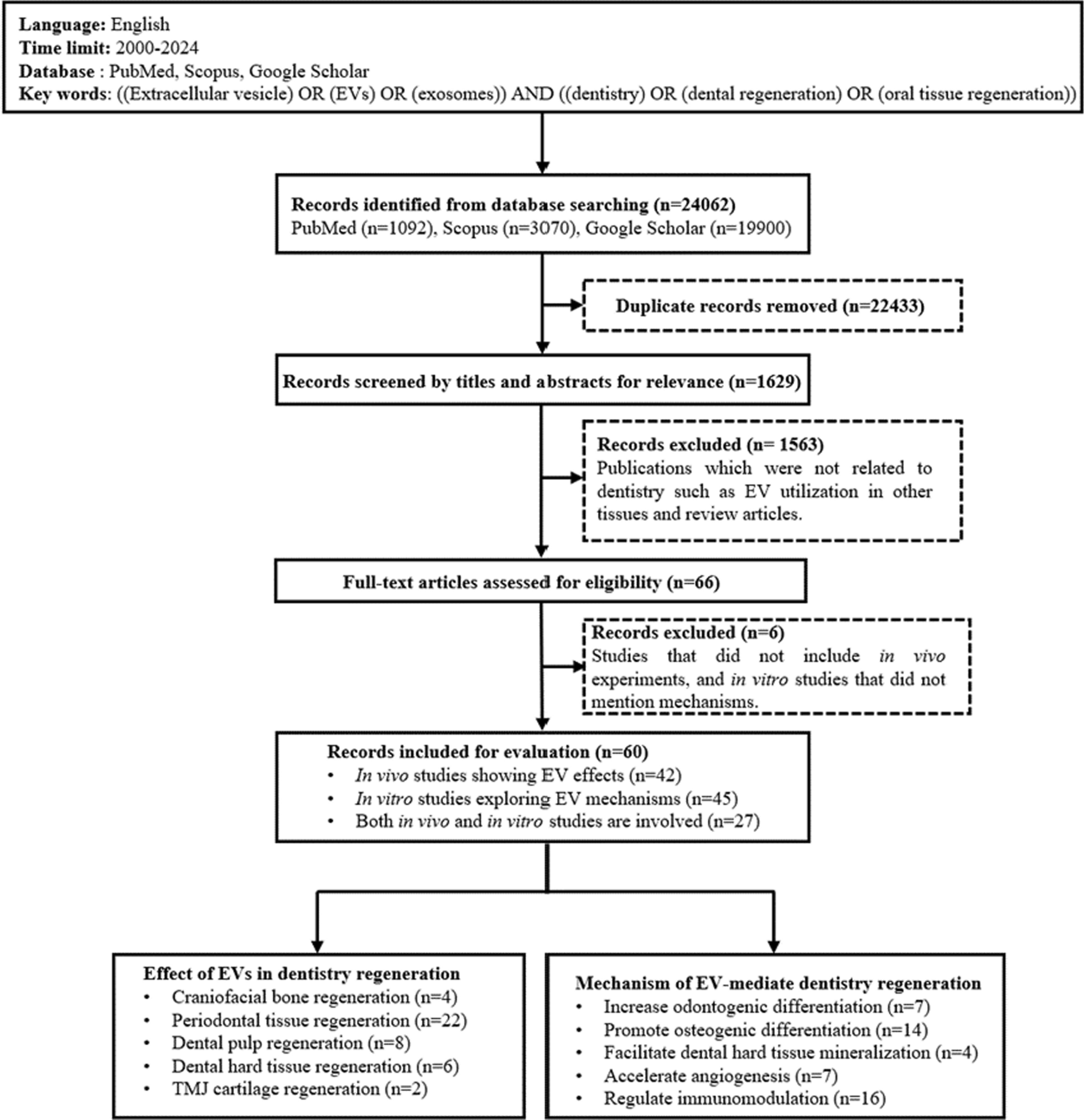
Sample collection
Informed consent was obtained from healthy human immunodeficiency virus (HIV)-negative mothers scheduled for caesarean section at 37 to 40 weeks gestation at a private hospital in Pretoria, South Africa. UCB was collected into bags containing citrate phosphate dextrose anticoagulant (Tianhe Pharmaceutical, China), stored at 4°C and used within 24 h. The HIV status of the mothers was obtained from their files or verbally during informed consent, and confirmed using the GeneXpert 1 System (Cepheid, USA) and Xpert® HIV-1 Qual or HIV-1 Qual XC cartridges (Cepheid, USA). Three individual donor samples were used for SR1 concentration optimisation, three samples for the 4-colour immunophenotype panel, and five samples for the 8-colour immunophenotype panel. For the gene expression, three pooled samples (each containing two UCB units) were used.
CD34+ HSPC enrichment
UCB was layered onto Histopaque®-1077 (Sigma-Aldrich, USA) in sterile 50 mL Falcon tubes at a 2:1 volume ratio and centrifuged for 30 min at 388 x g. The plasma fraction was aspirated, and the mononuclear cell (MNC) layer collected. Red blood cells were lysed using ammonium chloride (NH4Cl, pH 7.4) and the samples incubated at 4°C. Samples were centrifuged at 300 x g for 10 min at 4°C and NH4Cl was aspirated after which the cells were washed twice with TP buffer [phosphate buffered saline (PBS, pH 7.4), 10 µg/mL human albumin (Sigma-Aldrich, USA), and 2 mM ethylenediaminetetraacetic acid (EDTA)]. The supernatant was aspirated, and the cells were resuspended in up to 10 mL of TP buffer. A MNC aliquot was stained with CD45 (clone J33) FITC, CD34 (clone 581) PE and the viability dye, 7AAD (Stem-Kit™ HSPC enumeration kit, Beckman Coulter, Miami, USA) to identify viable CD34+ HSPCs. A second aliquot of the MNC suspension was stained with CD45 FITC (clone J33) and IsoClonic control PE and 7AAD (Stem-Kit™ HSPC enumeration kit, Beckman Coulter, Miami, USA) to determine the negative/positive boundary of CD34 expression. Viable CD34+ cells from the MNC fraction were sorted into 24-well plates using the BD FACSAria™ Fusion cell sorter (BD Biosciences, USA). Sorting purities of > 90% were achieved. Depending on the experiment, CD34+ HSPCs were sorted into serum-free expansion medium (StemSpan ACF/AOF; StemCell Technologies, Canada) or directly into lysis buffer (Qiagen, Germany).
Culture conditions
CD34+ HSPCs were cultured in 24-well plates (1 × 104 cells/well) in 1 mL StemSpan ACF/AOF medium supplemented with 2% penicillin (100 units/mL, GibcoBRL, USA), streptomycin (0.1 mg/mL, GibcoBRL, USA) and recombinant human growth factors, each at 100 ng/mL: SCF, TPO, FLT3L, granulocyte colony-stimulating factor (G-CSF) and IL-3 (Life Technologies, Thermo Fisher Scientific, USA). Cytokine combinations were optimised prior to starting these experiments (Additional file 1: Tables S1-2 and Figures S1-S6). Cultures were maintained at 37°C and 5% CO2 in a humidified atmosphere for 7–8 days. SR1 (StemCell Technologies, Canada) was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, USA) and further dilutions were made using StemSpan ACF/AOF. The following concentrations were tested to determine the optimal SR1 concentration for HSPC expansion: 0.25, 0.5, 0.75 and 1 µM. A vehicle control (VC) containing DMSO (0.01%) was included. Subsequent expansion, immunophenotype and gene expression experiments were performed using 1 µM SR1.
Flow cytometry
Two separate antibody panels, one with four and another with eight colours, were used to determine viability and CD34+ percentages on Day 0 (D0) and Day 7 (D7). The 4-colour immunophenotype panel was acquired using a 3-laser, 10-colour Gallios flow cytometer (Beckman Coulter, Miami, USA), while the subsequent larger 8-colour panel was acquired using a CytoFLEX flow cytometer (Beckman Coulter, Miami, USA). The D7 subsets were analysed in those cells expanded with (SR1) and without SR1 (VC).
The 4-colour immunophenotypic panel included the following anti-human monoclonal antibodies: Lin FITC (clones: CD3, UCHT1; CD14, HCD14; CD16, 3G8; CD19, HIB19; CD20, 2H7; CD56, HCD56; BioLegend, USA), CD34 PE-Cy7 (clone: 581; BioLegend, USA), CD38 APC-Cy7 (clone: HIT2; BioLegend, USA), CD133/2 PE (clone: 293C3; Myltenyi Biotec, Germany) and Mouse IgG2b PE isotype (clone: IS6-11E5.11; Myltenyi Biotec, Germany). The analysis was performed using either a Gallios flow cytometer or BD FACSAria Fusion cell sorter. Post-acquisition analyses were performed using Kaluza Analysis Software (version 2.1; Beckman Coulter, Miami, USA). The flow cytometric protocol and gating strategy are summarised in Additional file 2: Fig. S1. Fold increase in absolute cell numbers as well as percentage was used to determine cell sub-populations before and after a 7-day expansion. Fold increase refers to the factor by which the number of cells observed on D7 increased relative to the number of cells seeded on D0, and was calculated as follows:
$$\eqalign\text\text\text\text\:\text\text\text\text\text\text\:\text\text\:\text\text\text\text\text\:\text\text\:\text7-\:\text\text\text\text\text\:\text\text\text\text\text\text\:\text\text\:\text\text\text\text\text\:\text\text\text\text\text\text\:\text\text\:\text0\:\:}\text\text\text\text\:\text\text\text\text\text\text\:\text\text\:\text\text\text\text\text\:\text\text\text\text\text\text\:\text\text\:\text0}}$$
The 8-colour immunophenotype panel included the following anti-human monoclonal antibodies: Lin FITC (clones: CD3, UCHT1; CD14, HCD14; CD16, 3G8; CD19, HIB19; CD20, 2H7; CD56, HCD56; BioLegend, USA), CD34 APC AF700 (clone: 581; Beckman Coulter, Miami, USA), CD38 ECD (clone: LS198-4-3; Beckman Coulter, Miami, USA), CD133 PE-Violet 770 (clone: REA753; Miltenyi Biotec, Germany), CD117 PE (clone: 104D2; BioLegend, USA), CD90 BV510 (clone: 5E10; BioLegend, USA), CD45RA APC (clone: 2H4; Beckman Coulter, Miami, USA), CD49f SB780 (clone: G0H3; BioLegend, USA). Prior to determining the antibody volumes for the 8-colour panel, antibody titrations were performed and decisions on volumes were made based on the staining index [Stain Index = (Median of Positive – Median of Negative) / (Standard Deviation of Negative * 2)]. Post-acquisition analyses were performed using Kaluza Analysis Software (version 2.1; Beckman Coulter, Miami, USA) and Cytobank software (http://www.cytobank.org; Beckman Coulter, Miami, USA). The gating strategy employed prior to Cytobank analysis is summarised in Additional file 2: Fig. S2. Once uploaded into the Cytobank platform, data clean-up was performed using Peak Extraction and Cleaning Oriented Quality Control (PeacoQC) to identify and exclude any anomalous events. For the description of the D0 and D7 immunophenotype, all associated files were concatenated prior to Cytobank analysis. For statistical analysis, sample files were analysed individually and compared. Dimensionality reduction was performed using t-distributed stochastic neighbour embedding (tSNE) plots, after which a flow cytometry self-organising map (FlowSOM) was performed to determine statistically significant differences in populations in the D7 subsets (SR1 versus VC) using box plots and heat maps. Statistical analysis was performed using non-parametric Kruskal-Wallis and Mann-Whitney U tests where appropriate.
Side population (SP) analysis
SP analysis has proven to be a valuable tool to identify a population of immature HSPCs [33]. SP data was obtained from two different donors. Primitive (immature) stem cells preferentially efflux cytoplasmic lipophilic fluorescent dyes, such as the DNA binding dyes Vybrant® DyeCycle™ (VDC) Violet and Hoechst, resulting in a sub-population of cells displaying lower fluorescent intensity levels. The SP fraction (displayed as a percentage) was identified as cells with higher dye efflux ability and therefore cells with low/negative VDC Violet fluorescence compared to the rest of the population. SP analysis was performed after eight days in culture. Triplicate wells were pooled for SP analysis at a concentration of 103 viable cells/µL medium, in a total of 1 mL per tube. A Verapamil control (100 µM; Sigma-Aldrich, USA) was included for each condition. VDC Violet (5 µg/mL; Life Technologies, Thermo Fisher Scientific, USA) was added to all tubes and incubated at 37°C for 120 min, protected from light. Following incubation, cells were placed on ice and analysed on the BD FACSAria Fusion. Cells were stained with Annexin V FITC (Beckman Coulter, Miami, USA), 7AAD, CD34 PE-Cy7 and CD38 APC-Cy7 prior to analysis. The flow cytometric protocol and gating strategy are summarised in Additional file 1: Fig. S3.
RNA extractions, integrity and quality
The Qiagen RNeasy Micro Plus kit (Qiagen, Germany) was used to perform RNA extractions according to the manufacturer’s instructions. Cells (105) were sorted into 300 µL lysis buffer and were kept at 4°C throughout the sorting process. RNA was extracted from two aliquots of cells (105 each) on D0 and concentrated by vacuum centrifugation using a SpeedVac SVC-100 (Savant Instruments, USA). RNA integrity and quality were assessed using the TapeStation® 2200 (Agilent Technologies, USA), RNA ScreenTape® (Agilent Technologies, USA) and Sample Buffer Kit (Agilent Technologies, USA).
Gene expression
Gene expression analysis (50 ng total RNA in each sample) was performed using Affymetrix GeneChip® Human Gene 2.0 ST arrays (Affymetrix, USA) and Affymetrix GeneChip® WT PLUS Reagent Kit (Affymetrix, USA) according to the manufacturer’s manual. Purified, fragmented and labelled complementary RNA was added to the hybridization cocktail using Hybridization Wash and Stain Kit (Affymetrix, USA). The hybridization cocktail was hybridized to Affymetrix GeneChip® Human Gene 2.0 ST arrays for 17 h. The GeneChips were placed in an Affymetrix GeneChip® Hybridization Oven-645 (Affymetrix, USA) at 45°C rotating at 60 rpm. The hybridized chips were washed and stained in an Affymetrix GeneChip® Fluidics Station-450Dx (Affymetrix, USA) before scanning using an Affymetrix GeneChip® Scanner-7G (Affymetrix). Analyses were performed using the Affymetrix Transcriptome Analysis Console™ (TAC) Software 4.0 (Affymetrix, USA). Genes with a fold change ≥ 2.5 and ≤ -2.5 and (P < 0.05) were considered to be differentially expressed. The microarray data files of this study have been submitted to NCBI GEO (Gene Expression Omnibus) with accession number GSE146810. The Database for Annotation, Visualization, and Integrated Discovery (DAVID) analysis tool (https://david.ncifcrf.gov) was used to classify differentially expressed genes into biological processes [34].
Statistical analysis
Experiments were performed in triplicate using two to eight independent UCB samples. Values represent mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism (Version 10.0.2) as well as the Cytobank platform. A non-parametric one- or two-way analysis of variance (ANOVA), a Kruskal-Wallis, a Mann-Whitney U test and a Dunn’s Multiple Comparison Test were performed where appropriate to determine statistical significance (P < 0.05).









留言 (0)