
記住我








Between November 2018 and November 2022, we collected 1062 CSF samples from adult and pediatric patients seeking oncology care or molecular pathology consultation at Memorial Sloan Kettering Cancer Center (MSK); 55 CSF samples were excluded from further genomic profiling for logistic or administrative reasons. The final sequenced cohort included 1007 CSF samples from 711 patients (Supplementary Fig. 1a-b). We collected multiple CSF samples in 150/711 (21.1%) across their disease course (median: 2, range: 2–12 samples per patient) (Supplementary Fig. 1c). All CSF samples were processed and sequenced upon receipt in the clinical laboratory, in parallel with the corresponding matched normal (blood) sample.
Our patient cohort included patients with over 90 distinct tumor types of primary CNS and metastatic origin, including lung cancer (n = 188), breast cancer (n = 150) and gliomas (n = 148) as the most common broad categories (Supplementary Table 1). 85/1007 (8.5%) of the CSF samples were collected from patients who did not have any clinically documented evidence of CNS involvement by cancer. The median follow-up following CSF collection was 240 days (IQR: 112–483 days).
Landscape of somatic genomic alterations detected in CSF922/1007 (91.5%) CSF samples were collected from patients with clinical CNS disease; of these, 53% (489/922) harbored at least one somatic genetic alteration. These samples were categorized as ctDNA positive (ctDNA +).
By contrast, all samples collected from patients without any clinically documented CNS involvement by cancer (85/85) were negative for genetic alterations (specificity = 100%).
A total of 7110 somatic alterations (3944 somatic non-synonymous mutations, 2980 somatic copy number alterations, and 186 structural rearrangements) were detected across the 489 CSF ctDNA + samples. The number of mutations and variant allele frequencies (VAF) varied across ctDNA + samples, with a median of 4 mutations per sample (IQR: 2–8, range: 1–415) and VAFs ranging from 1 to 100% (median VAF: 38.7%, IQR: 23.2–51.1%). Tumor types varied by rates of ctDNA positivity, with GI cancers having the greatest proportion of CSF samples harboring at least one genetic alteration and embryonal tumors having the fewest. Likewise, VAF varied across the different tumor types with GI cancers having the greatest median VAF compared with other tumor types, possibly reflecting different rates of ctDNA shed across tumor types and the varying propensities for leptomeningeal versus parenchymal-only disease (Supplementary Fig. 2).
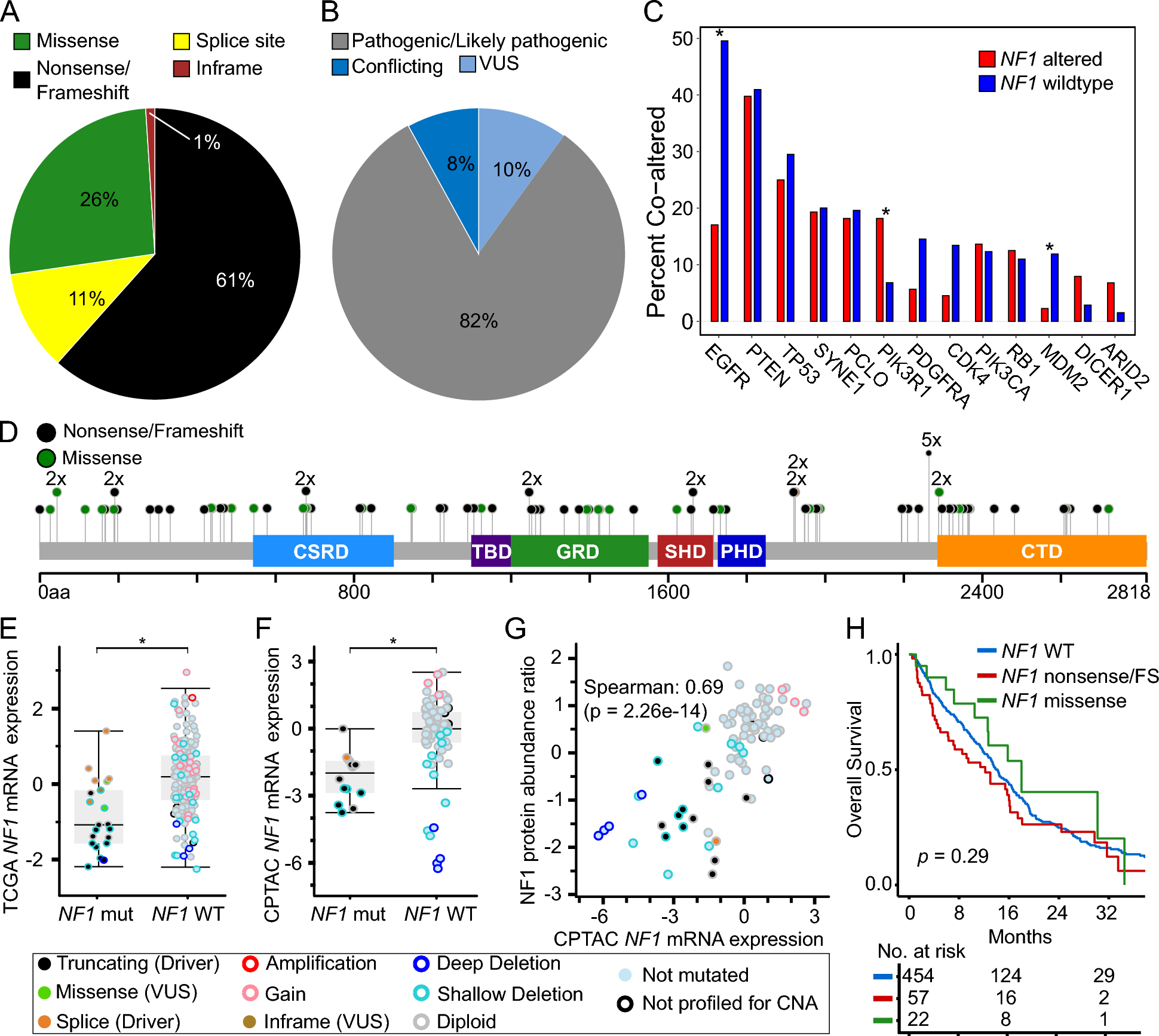
We observed the full spectrum of genetic alterations in CSF ctDNA. TP53 was the most frequently altered gene across all tumor types (n = 242, 49% of 489 ctDNA + samples). Other commonly mutated genes were consistent with the expected landscapes of individual tumor types. For instance, both mutation and high amplification could be detected in EGFR, MET, and ERBB2 in lung cancer, as well as mutations in KRAS, BRAF, STK11, KEAP1 and many others. Mutations in PIK3CA p.E545K and amplification in ERBB2 were common in breast cancers, BRAF p.V600E in metastatic melanoma and IDH1 p.R132H in IDH-mutant gliomas (Fig. 1a).
Fig. 1
Genomic alterations detected in CSF-ctDNA. a Oncoplot of the most frequently altered genes, stratified by broad tumor categories. Each column represents an individual sample. Plot includes non-synonymous mutations, indels, copy number alterations and structural variants. Upper bars depict TMB levels; the dashed green line indicates a TMB of 10 muts/Mb. Lowest track indicates the tumor category. Multi_Hit refers to those genes that were mutated more than once in the same sample. b, c Circos plot of the 186 structural variants identified. Arrows highlight select recurrent alterations with the most clinical relevance, further stratified by number and clinical implication as diagnostic or therapeutic. d Distribution of observed mutation rates across CSF samples sequenced; a threshold of 13.8 mutations/Mb was considered indicative of high mutation burden based on historical analysis of 10,000 tumor samples by MSK-IMPACT testing (left). Dominant mutation signatures identified in cases with high mutation burden. The percent of cases harboring a dominant mutation signature is shown for each broad tumor category (right panel). MMR: Mismatch repair deficiency; UV: Ultraviolet light; TMZ: Temozolomide
Based on assay design and coverage of key intronic regions of the genome, we were also able to detect a broad range of clinically relevant fusions, such as EML4::ALK, RET, and ROS1 rearrangements with diverse gene partners in lung carcinomas, BRAF::KIAA1549 and EGFRvIII alterations in gliomas (Fig. 1b and c).
Beyond the individual somatic alterations, we interrogated CSF samples with the highest tumor mutation burden (≥ 13.8 mutations/Mb, n = 35) for the presence of mutational signatures and identified signatures related to prior exposure to ultraviolet (UV, n = 3), APOBEC (n = 7), smoking (n = 8), and temozolomide (n = 2) in metastatic cutaneous melanomas, breast cancers, lung cancers, and gliomas, respectively (Fig. 1d). The threshold of 13.8 mutations/Mb was chosen as indicative of high mutation burden, based on historical analysis of 10,000 tumor samples by MSK-IMPACT testing [22].
Clinical relevance of CSF-ctDNA positivityTo assess the clinical relevance of somatic variants in CSF, we annotated each of the alterations detected by their level of clinical actionability according to the OncoKB (https://www.oncokb.org/) precision oncology knowledge base [7]. OncoKB was recognized by the Food and Drug Administration as a tumor mutation database that provides information about the biological and clinical implications of over 5,000 cancer gene alterations. Level 1 alterations in OncoKB are defined as FDA-recognized biomarkers predictive of response to an FDA-approved drug in this indication. Level 2 alterations are standard care biomarkers recommended by the NCCN or other professional guidelines predictive of response to an FDA-approved drug in this indication. Level 3A alterations require compelling clinical evidence to support the biomarker as being predictive of response to a drug in this indication.
Across the 489 ctDNA + samples, 248 (50.7%) had a level 1 OncoKB actionable alteration. Lung carcinomas had the highest level 1 actionability in the cohort, consistent with the number of precision oncology drugs currently available in lung cancer. In this subset, we also detected the highest number of alterations predictive of therapeutic resistance (OncoKB R1 and R2) which informed further patient management. Among other malignancies, the OncoKB levels of actionability in the CSF were broadly comparable by cancer type to those found in tissues sequenced by MSK-IMPACT as part of the AACR GENIE cohort (n = 47,271) (Fig. 2a).
Fig. 2
Clinical Relevance of CSF-ctDNA positivity. a Alterations detected in cfDNA from CSF were annotated and stratified by their level of clinical actionability according to the OncoKB precision oncology knowledge base. The proportions were compared to the AACR-Genie MSK cohort of solid tumor (n = 47,271). There is a relative enrichment for level 1 alterations due to the use of the assay for monitoring of patients on targeted therapies. b Survival curves showing that detection of ctDNA in CSF is associated with lower survival probability
In addition to the assessment of therapeutic actionability, genomic profiles provided pivotal information for tumor subclassifications. For both primary and suspected metastatic lesions, profiling established clonal relatedness to a known malignancy and, in some cases, informed the presence of a previously unsuspected tumor (Supplementary Figs. 3 and 4).
Notably, given the relative purity of ctDNA and the high number of genetic alterations that could be detected in some cases, the assessment of mutational signatures assisted in the determination or confirmation of the primary tumor site, such as UV signatures or smoking signatures in suspected metastatic cutaneous melanoma or lung adenocarcinoma, respectively (Supplementary Fig. 5).
We determined the relationship between ctDNA positivity and overall survival (OS) in our cohort. Across all cancer types, detection of a genetic alteration in the CSF was associated with a three-fold increased risk of death (HR: 3.23, 95% CI: 2.58–4.05, P < 0.001). Median survival was 854 days shorter in patients with CSF positivity than otherwise (detected alteration: 235 days (95%CI: 177–272 days); undetected alteration: 1089 days (95%CI: 796 days-not reached) (Fig. 2b) (Supplementary Table 2). The association between shortened OS and CSF positivity was seen across all tumor subtypes (except embryonal and GI cancers) with no statistically significant heterogeneity (P = 0.13, Supplementary Fig. 6).
CSF sampling in patients with metastatic lung cancerPatients with lung cancer represented the largest subgroup of patients in our dataset and the subgroup of patients with the most FDA-approved genotype-directed therapies. This allowed a closer look into the role of CSF sampling as a tool for CNS assessment and disease monitoring.
Among the lung cancer patients those with parenchymal brain metastases and additional evidence of leptomeningeal involvement (defined by positive cytology, positive circulating tumor cells ≥ 3, or radiographic leptomeningeal spread as called by the formal clinical radiology report) were more likely to have positive CSF ctDNA than those with parenchymal brain metastases in the absence of leptomeningeal involvement (OR: 20.17; CI: 9.65–42.16; p < 0.0001) (Supplementary Table 3, Additional File 1). After excluding cytology samples that were reported as atypical or suspicious by pathology (n = 33), detection of a genetic alteration in the CSF had greater sensitivity than positive cytology for the presence of leptomeningeal disease (sensitivity: 85.4% vs. 61.7%) and greater negative predictive value (80% vs. 66%). Since ctDNA positivity also occurred in some patients with parenchymal-only disease, this finding was not entirely specific for leptomeningeal disease (specificity: 78.7%, positive predictive value: 84.4%).
In lung cancer patients, the driver alterations initially detected in the tumor tissue were universally detected in the CSF (Fig. 3a). Among lung cancer patients with EGFR sensitizing mutations and on therapy, CSF sequencing demonstrated the emergence of gatekeeper mutations associated with acquired resistance, including EGFR p.T790M, p.C797S, p.L792H, p.L718Q, p.L718V and p.G724S. Other acquired alterations included amplifications in MET, and EGFR, and off target alterations in BRAF (fusion), KRAS, PIK3CA and others (Fig. 3b). Some patients harbored several alterations as exemplified by a patient with EGFR-mutant non-small cell lung carcinoma where repeated CSF sequencing identified the emergence of multiple different EGFR mutations in response to first and third-generation EGFR inhibitors (Fig. 3c). Among patients with ALK fusions and MET exon 14 skipping mutations, emergence of additional ALK mutations (p.G1202R and p.G1269A) and MET alterations (p.Y1230N) were also detected upon progression on targeted therapy (Supplementary Fig. 7).
NSCLC patients with positive CSF-ctDNA had significantly shorter survival following CSF collection than NSCLC patients with negative CSF-ctDNA (Fig. 3d).
Fig. 3
CSF sampling in NSCLC. a 77 samples with actionable driver alterations detected in CSF were compared to results from prior tissue biopsies. Driver alterations initially detected in the tumor tissue were universally detected in the CSF. Each column represents 1 patient. Blue boxes designate those samples where both CSF and tissue sequencing demonstrated the same driver alteration. In two cases, the mutation detected in the CSF was distinct form the one detected in the tumor. In both cases, retrospective review demonstrated the presence of multifocal lung disease with the metastasis representing a separate primary that was not previously sequenced. b Among patients with EGFR sensitizing mutations, sequencing of CSF from 28 patients detected several additional alterations associated with secondary resistance, including mutations in EGFR, and alterations in other genes (MET, PIK3CA, BRAF). c Representative case of a patient with EGFR mutated lung adenocarcinoma and monitoring starting at the time of suspected CNS metastasis. 7 CSF samples are obtained demonstrating the gradual emergence of several resistance mechanisms associated with treatment with EGFR inhibitors (T790M, L718V and L718Q. The table displays the mutations detected in each sample sequenced, along with the corresponding VAF’s (%), highlighted according to the color scale (bottom left). Lowest track denotes the classification of the EGFR mutations as sensitizing (L1, green) or associated with acquired resistance (R1 standard care resistance; R2 investigational resistance, red) according to OncoKB. L4 (dark gray) denotes an alteration with compelling biological evidence that supports the biomarker as being predictive of response to a drug. d Survival curves for NSCLC patients demonstrate that detection of ctDNA in CSF is associated with lower survival probability
Comparison of CSF ctDNA with plasma and tumor tissueSeveral patients in our cohort had undergone a tumor biopsy or plasma collection within 90 days of the CSF collection. This provided an opportunity to compare the representation of the cancer genome in CSF compared to tumor tissue or blood.
Correlations between somatic alterations in CSF ctDNA and tumor DNA were possible for 56 pairs (55 patients). Overall, we detected 999 alterations in tumor and CSF from these patients. 434/999 (43%) alterations were shared between tumor and CSF, 273/999 (27%) were private to the CSF and 292/999 (29%) were private to the tumor biopsy (Supplementary Fig. 8a). The frequency of shared CSF/tissue alterations was considerably higher (41/53 = 77%) than private alterations to CSF or tissue for the most clinically relevant alterations (OncoKB levels 1 to 3A) (Supplementary Fig. 8b). A comparison of VAFs for shared mutations revealed significantly higher levels in ctDNA from CSF (median VAF: 32%, IQR: 27%) compared to the tumor tissue (median VAF: 24%, IQR: 33%), despite routine enrichment by manual macro-dissection in solid tumor samples where necessary (P < 0.01, Mann–Whitney U test) (Supplementary Fig. 8c-d). Measurements of tumor mutation burden (TMB) in tumor tissue and CSF corresponded closely with each other (r = 0.81, P < 0.001, Spearman’s rank correlation) (Supplementary Fig. 8e.
Comparisons of somatic alterations detected in ctDNA from CSF versus plasma was performed on 31 patients and focused on the somatic mutations targeted by both assays (MSK-IMPACT and MSK-ACCESS). Over half of the total alterations were shared between plasma and CSF (77/142, 54%) (Supplementary Fig. 9a), and included the majority of clinically relevant mutations (24/32 mutations (75%), OncoKB levels 1 to 3A) (Supplementary Fig. 9b). Compared to the alterations identified in plasma, mutations detected in CSF cfDNA were identified at significantly higher VAFs (CSF: median VAF = 36.4%, IQR = 34.3% vs. Plasma: median VAF = 2.3%, IQR = 10.7%, P < 0.01, Mann–Whitney U test), likely reflecting greater dilution of tumor-derived DNA by non-neoplastic DNA in blood (Supplementary Fig. 9c).
Determinants of CSF-ctDNA positivityThe rates of CSF ctDNA positivity among patients with primary CNS tumors versus CNS metastasis were compared, excluding the small subset of cases with tumors of unknown primary origin (n = 26). Samples from patients with CNS metastasis were more likely to be ctDNA positive (OR = 2.60, CI 1.96–3.46, P < 0.001, Fisher’s exact test) (Fig. 4a).
Fig. 4
Pre-analytic factors associated with CSF-ctDNA positivity. a Stratification of samples based on disease type (primary CNS tumor vs metastasis) shows that that metastatic tumors have higher rates of ctDNA positivity than primary tumors. b Samples are stratified by the volume of CSF received for testing. While genetic alterations could be detected even in the context of very low volume samples, the rate of positivity was critically impacted for those samples below 2 ml. These samples were associated with rates of ctDNA positivity between 8 and 20%. The rate of positivity increases as volumes reach 5 ml and above. Yellow bars indicate the proportion of samples that are ctDNA positive. Blue bars indicate those that are ctDNA negative (no detected genetic alterations). c Analysis of rates of positivity for samples according to time to extraction. Across the entire cohort, increased time to extraction was not associated with increased proportion of ctDNA negative samples. Samples extracted outside the stability criteria of STREK tubes (> 14 days) constituted a very small proportion of the samples–this very small subset demonstrated a drop in the rate of positivity compared to those extracted before 14 days but the number was too low for a conclusive analysis. d, e Comparisons of total DNA yields and sequencing coverages between ctDNA + and ctDNA- samples. Overall, the proportion of ctDNA + samples increased with higher DNA yield and, consequently, higher sample coverages. f A broad range of coverages are found across CSF samples. Top graph shows the range of coverages across the entire cohort. Lower panel and insert (right) display the zoomed views of the samples with lowest coverages. Despite the low coverages, detection of genetic alterations remains possible in many cases below 50× due to the high proportion of ctDNA in CSF samples (not diluted by cfDNA from hematopoietic components). Higher proportion of samples have ctDNA detected when sample coverage increases
While our standard recommendation for sample collection was 10 mL of CSF in Streck tubes, we received highly variable sample volumes (Fig. 4b). The median CSF volume submitted was 5.5 mL (range 0.4–30 mL, IQR: 4.5 mL). Most samples (99.0%) were received in Streck BCT tubes; 1.0% arrived in sterile containers and were immediately transferred to Streck tubes in our lab; 99.2% of samples were processed within the established stability window of the Streck collection tubes (≤ 14 days). Delays in processing reflected lags in shipping and transport when samples were procured at outside hospitals; 45 samples (4.5%) were flagged due to deviations in quality control (received in non-Streck tubes, blood-tinged samples or frozen Streck tube collections).
In this cohort, delays in CSF DNA extraction did not adversely affect the rate of CSF ctDNA positivity (r = 0.27, P = 0.29, Spearman’s rank correlation) (Fig. 4c). However, the proportion of ctDNA in the samples decreased with increasing time to extraction (r = − 0.07, P = 0.03, Spearman’s rank correlation) beyond 7 days, likely related to dilution associated with gDNA released from cells when CSF is not separated. For the small subset of samples submitted in non-Streck tubes and those visually bloody, we observed significantly lower VAF’s compared to Streck collections (median VAF = 23.5% vs. 38.9%, respectively, P < 0.001, Mann–Whitney U test).
While genetic alterations could be detected even in the context of very low volume samples, the rate of positivity was critically impacted for those samples below 2 ml. For these low volumes, samples from patients with CNS disease that were appropriately collected in Streck tubes were associated with rates of ctDNA positivity as low as 8.3% and 20.0% for primary and metastatic CNS tumors, respectively. The rate of positivity increased to 37.8% and 68.6% for primary and metastatic tumors, respectively, as volumes increased to 5 ml. We noticed no significant improvements in ctDNA positivity rates with volumes of 10 ml and above.
The median cfDNA yield was 0.29 ng (range: 0 to 872.5 ng, IQR: 0.11–2.19 ng). All samples were sequenced regardless of the DNA quantity recovered. The median sequencing coverage was 65X (range: 0- 2735X; IQR: 16-295X); 457 samples had coverages below 50X, corresponding to those with lowest cfDNA yields (median DNA yield 0.12 ng [IQR: 0.08–0.19 ng] vs. 1.69 ng [IQR: 0.45–6.03 ng] in samples with greater than 50X coverage).
Comparisons of DNA yields, and coverages between ctDNA + and ctDNA- samples are summarized in Fig. 4d–f and Supplementary Table 4. Overall, the proportion of ctDNA + samples increased with higher sample coverages, while sample coverage correlated strongly with DNA yield (r = 0.79, P < 0.001, Spearman’s rank correlation).
留言 (0)