Participants
The recruitment procedure, eligibility criteria, and enrollment of the LLFS participants have been previously described (Newman et al. 2011). We used data from the first clinical exam, which started in 2006 and recruited 4953 individuals from 539 families. Across 11 studied traits, the participants ranged from n = 2528 to 4166 for GWAS, n = 595 to 1200 for gene expression level–trait association, and n = 2528 to 4166 for rare-variant analysis. Descriptive statistics for all the traits and covariates can be found in File S3. The number of participants in each analysis depended on the number of participants with data for the trait, microarray genotypes for GWAS, whole genome sequencing for rare-variant analysis, and RNA-Seq for TWAS.
Cardiovascular-related traits
We used trait values from the first clinical exam. BMI was calculated as weight (kg)/height (m)2, and waist as the average of three abdominal circumference measurements in cm. Pulse was calculated as the average of three measurements of the sitting pulse. FEV1 and FVC were measured in a portable spirometer (EasyOne, NDD Medical Technologies, Andover, MA), as previously reported (Newman et al. 2011). High-density lipoprotein (HDL), low-density lipoprotein (LDL), triglyceride (TG), and total cholesterol (TC) were assessed and analyzed by the LLFS central laboratory based at the University of Minnesota, as previously reported (Newman et al. 2011). Participants were excluded if fasting < 8 h for LDL, TG, and TC. Ankle-brachial index (ABI) was derived as the average of the right and left ankle-arm blood pressure ratios. We excluded participants with non-compressible arteries (ABI > = 1.4). For all analyses, each of the traits was adjusted for age, sex, field center, and square of the age. Waist and pulse were additionally adjusted for BMI. FEV1, FVC, and FEV1/FVC were also adjusted for height and smoking. LDL and TC were also adjusted for statin use, and TG was log-transformed. All traits were also adjusted for the top 10 genetic principal components stepwise. The trait residuals unexplained by the covariates were used for GWAS, TWAS, and RVA. After covariate adjustments, all trait residuals were inverse normal transformed.
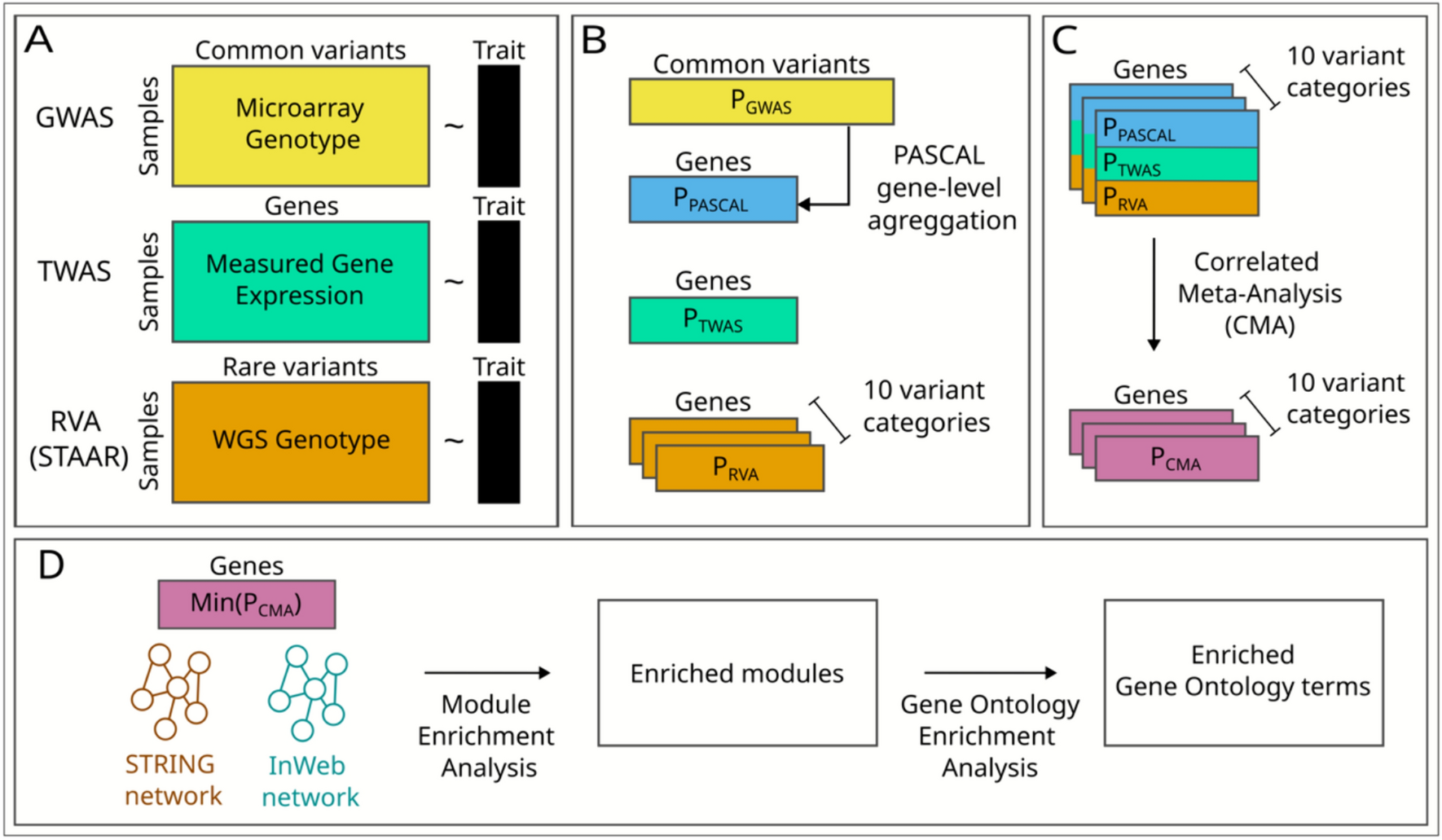
GWAS and gene level aggregation of GWAS results
GWAS SNP-chip data for the LLFS participants were produced using Illumina 2.5 million HumanOmni array. Genotypes were called using Bead Studio. SNPs were removed if their call rate was less than 98%, if their allele frequency in the LLFS population was < 1% or > 99%, if they had an allelic mismatch with 1000 Genomes Project (1000Gp3v5), or if they displayed excess heterozygosity relative to Hardy Weinburg Equilibrium (p < 1E–6). A single-SNP association test was done for all SNPs passing the quality filter by using a linear mixed model. Family relatedness was accounted for using a pedigree-based kinship matrix, and an additive genetic model was assumed. The SNP-level summary statistics from GWAS for SNPs with minor allele frequency > = 5% were input to PASCAL (Lamparter et al. 2016). The SNPs were assigned to a gene if they lay within 50 kb of the gene body. PASCAL uses the sum of chi-squared statistics to calculate a gene-level p-value. Document S1 describes the GWAS and gene level aggregation process for the FHS population.
Gene-expression to trait association (TWAS)
The RNA extraction and sequencing were carried out by the McDonnell Genome Institute at Washington University (MGI). Total RNA was extracted from PAXgene™ Blood RNA tubes using the Qiagen PreAnalytiX PAXgene Blood miRNA Kit (Qiagen, Valencia, CA). The Qiagen QIAcube extraction robot performed the extraction according to the company's protocol. The RNA-Seq data were processed with the nf-core/RNASeq pipeline version 3.3 using STAR/RSEM and otherwise default settings (https://zenodo.org/records/5146005). RNA-Seq on whole blood samples from the LLFS participants in the first clinical visit was used for the analysis. Genes with less than three counts per million in greater than 98.5% of samples were filtered out from the analysis. Samples with greater than 8% of reads in intergenic regions were also filtered out. The resulting set were transformed using DESeq2’s (Love et al. 2014) variance stabilizing transform (VST) function. The VST transformed gene expression levels were adjusted for base covariates: age, age squared, sex, field center, percent of reads mapping to intergenic sequence, and the counts of red blood cells, white blood cells, platelets, monocytes, and neutrophils. The gene expression level was also adjusted stepwise for the RNA-seq batch and the top 10 principal components of gene expression. For each trait, the adjusted gene expression residuals were used as predictors, and the trait residuals after adjustment were used as a response variable in a linear mixed model implemented in MMAP (O’Connell 2017). A kinship matrix generated by MMAP from the LLFS pedigree was used to account for family relatedness. For traits with genomic inflation factor (GIF) > 1.1, the p-values were adjusted using BACON (van Iterson et al. 2017). The same RNA-Seq processing steps were implemented for replication in the FHS dataset.
Rare-variant analysis (RVA) using STAAR
LLFS Whole Genome Sequence (WGS) was produced by MGI using 150 bp Illumina reads. Variant calls with read depth less than 20 or greater than 300 were set to missing. Variants with call rate < 90% and those with excess heterozygosity (p < 1E–6) were excluded from the analysis. Missing genotype calls in the LLFS cohort were filled in using the call with the highest phred-scale likelihood from GATK. Bi-allelic SNVs with MAF < 5% and passing the above quality filters were input to STAAR (Li et al. 2020) for variant set association tests using SKAT (Wu et al. 2011). We also employed burden testing (Morgenthaler and Thilly 2007; Li and Leal 2008; Madsen and Browning 2009; Morris and Zeggini 2010) and Aggregate Cauchy Association Test (ACAT) (Liu et al. 2019; Liu and Xie 2020) as implemented in the STAAR framework. However, the resulting p-value distributions from these tests displayed a U-shaped pattern, deviating from the expected uniform distribution under the null hypothesis so we did not use them.
For each gene, variants are split into 10 functional categories, and an omnibus association test is performed for each category of each gene separately, resulting in 10 p-values per gene. Within each category, variants are weighted by functional annotations from the FAVOR database (Zhou et al. 2023), which is curated by the TOPMed Consortium. The annotations describe various aspects of variant function, and include conservation scores, epigenetic measurements, protein function scores and metrics for relative pathogenicity. The individual annotation scores for each variant are integrated into a single variant-specific weight by calculating the first principal component of the standardized annotation scores, and then transforming the principal component into a Phred-scale score (Li et al. 2020). The 10 functional categories include synonymous, missense, putative loss of function (plof), promoter CAGE, promoter DHS, enhancer CAGE, enhancer DHS, upstream, downstream, and untranslated region (UTR) (Li et al. 2020; Zhou et al. 2023). A minimum of 2 variants is required in a category to perform a SKAT test. Document S1 describes the WGS data processing steps for the FHS population.
Correlated meta-analysis (CMA)
CMA (Province and Borecki 2013) combined gene p-values from GWAS (after aggregation by PASCAL), TWAS, and RVA while preventing Type I errors by accounting for dependencies between individual analyses under the null as described (Province and Borecki 2013; Feitosa et al. 2022, 2024). CMA uses the multivariate normal distribution to integrate GWAS, TWAS, and RVA p-values. The individual p-values are first converted to corresponding z-scores which are assumed to have a multivariate normal distribution. The combined p-values are calculated using the standardized z-scores. This calculation is based on the sum of the z-scores and its standard deviation. The standard deviation is determined based on an empirically estimated covariance matrix, which represents the correlation between the individual studies. GWAS, TWAS, and RVA were performed on overlapping individuals from LLFS’s first clinical visit. Furthermore, genetic variants affect gene expression. Therefore, each pair of inputs to CMA may be correlated. Since STAAR outputs 10 p-values per gene, one for each category, we ran CMA 10 times resulting in 10 p-values per gene. To account for multiple testing using Bonferroni correction and identify significant genes following CMA, the number of tests is aggregated across all 10 CMA runs. If a gene meets the Bonferroni significance threshold for any category after adjusting for the total number of tests, it is considered significant after CMA. The significance threshold used for GWAS, TWAS, RVA, and CMA for LLFS can be found in Table S4.
Module enrichment analysis and Gene Ontology (GO) over-representation analysis
We started with modules (highly connected subnetworks) from two protein–protein interaction (PPI) networks, the STRING functional PPI network (Szklarczyk et al. 2015) and the InWeb physical PPI network (Li et al. 2017) which were identified by the best-performing module identification methods from a DREAM challenge (Choobdar et al. 2019): random walk algorithm R1 for STRING and modularity optimization algorithm M2 for InWeb. These modules and the gene-level p-values were input to PASCAL’s module enrichment algorithm (Lamparter et al. 2016). Genes with p-values from only one of the three sources (GWAS, TWAS, or RVA) were removed from the modules. The module enrichment p-values from PASCAL were corrected for the total number of modules tested using Bonferroni correction. GO over-representation analysis was done on the set of genes in each enriched module by using WebGestaltR package (version: 0.4.6) with the following configuration: (organism: hsapiens, method: ORA, enrichDatabase: GO Biological Process, FDRMethod: BH, FDRThreshold = 0.05) (Wang et al. 2017). The affinity propagation feature in WebGestaltR was used to eliminate GO biological processes with highly overlapping member genes.
Framingham Heart Study (FHS) replication
FHS is a multi-generational study to identify genetic and environmental factors affecting cardiovascular and other diseases (Splansky et al. 2007; Kannel et al. 1979). We used the data on the FHS participants from grandchildren and offspring spouse generation who attended examination 2 for replication purposes (Splansky et al. 2007; Kannel et al. 1979). Across 11 studied traits, the participants ranged from n = 2512–3341 for GWAS, n = 1080–1380 for TWAS, and n = 921–1233 for rare-variant analysis. Descriptive statistics for all the traits and covariates can be found in File S3. We use the same pipeline described in Fig. 1 to replicate the LLFS results in the FHS population. Replication analysis was done on genes that were significant in LLFS by CMA or by any of the CMA inputs: TWAS, GWAS, or RVA. For each trait, a gene is replicated if it meets the Bonferroni significance threshold, which is adjusted for the number of genes that were significant in the LLFS population in GWAS and TWAS, or for the number of gene-category pairs of significant genes in CMA and STAAR. A module is replicated if it is significantly enriched after applying Bonferroni correction based on the number of significantly enriched modules across all traits in the LLFS population.
GWAS Catalog search
We used the NHGRI-EBI GWAS Catalog database (version: v1.0.2-associations_e109) (Sollis et al. 2023) to check if the gene-trait associations with suggestive/significant signals from GWAS, TWAS, RVA, and CMA have a previously known trait-associated genome-wide significant variant within 50 kb of the gene body. Genes matching this criterion are designated as “previously associated in GWAS Catalog” throughout the paper. It is important to note that the presence of previously known trait-associated variants in a 50 kb region around the trait-associated gene’s body does not necessarily establish a causal role for the gene on the trait. However, we use this broad criterion to ensure that we classify genes with any hint of previous implication as “previously associated,” minimizing the risk of incorrectly classifying them as novel findings.








留言 (0)