
記住我
Alzheimer’s Disease (AD) comprises approximately 60–70% of dementia cases and can be sub-categorized based on age of onset and inheritance pattern (WHO, 2023). Early-and late-onset forms of AD are defined as whether disease manifested before or after age 65, respectively (Reitz et al., 2020). Mendelian inheritance of AD is characterized by fully penetrant autosomal dominant inheritance of genes like mutated amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin (PSEN2), though it has also been used to describe AD patients with a family history of the disease (Reitz et al., 2020). Sporadic inheritance is characterized by inheritance patterns that are not obviously autosomal dominant or have a highly variable age at onset (Reitz et al., 2020). AD can be clinically described as “positive” brain lesions of amyloid plaques, tau tangles, and glial inflammatory responses and “negative” brain lesions of neuronal and synaptic loss (Serrano-Pozo et al., 2011). These “negative” lesions, especially synaptic loss, closely parallel cognitive decline (Serrano-Pozo et al., 2011). Neuroinflammation, localized to “positive” lesions, plays a significant role in both the response to and the perpetuation of disease processes, thereby acting as a “double-edged sword” (Swanson et al., 2018).
Microglia are brain-resident immune cells that play pivotal roles in neuroinflammation in AD (O’Connor and Nissen, 2023). In early disease stages, microglia are efficient at clearing amyloid plaque; however, as disease progresses, microglia transition from an “acute” to a “chronic” inflammatory phenotype often referred to as neurodegenerative microglia (MGnD) or disease-associated microglia (DAM) that are less effective at amyloid removal (O’Connor and Nissen, 2023; Kinney et al., 2018; Poppell et al., 2023; Ocañas et al., 2023). These MGnD/DAM microglia are thought to be responsible for damaging inflammation resulting in neuronal death (O’Connor and Nissen, 2023; Kinney et al., 2018; Poppell et al., 2023). Circulating peripheral immune cells contribute to this damaging inflammatory environment through cytokine secretion, creating a positive feedback loop with the microglia.
One group of these peripherally derived cytokines that act on microglia is the common cytokine receptor γ chain family, which includes the interleukins IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. These cytokines bind heterodimer receptors to activate three major signaling pathways: mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and Janus kinase/signal transducers and activators of transcription (JAK/STAT). This perspective focuses on the effect these cytokine receptors have on microglia, as they are upstream of signaling pathways implicated in AD-related neuroinflammation and occur in the immune cell considered most responsible for the inflammatory response (Table 1).
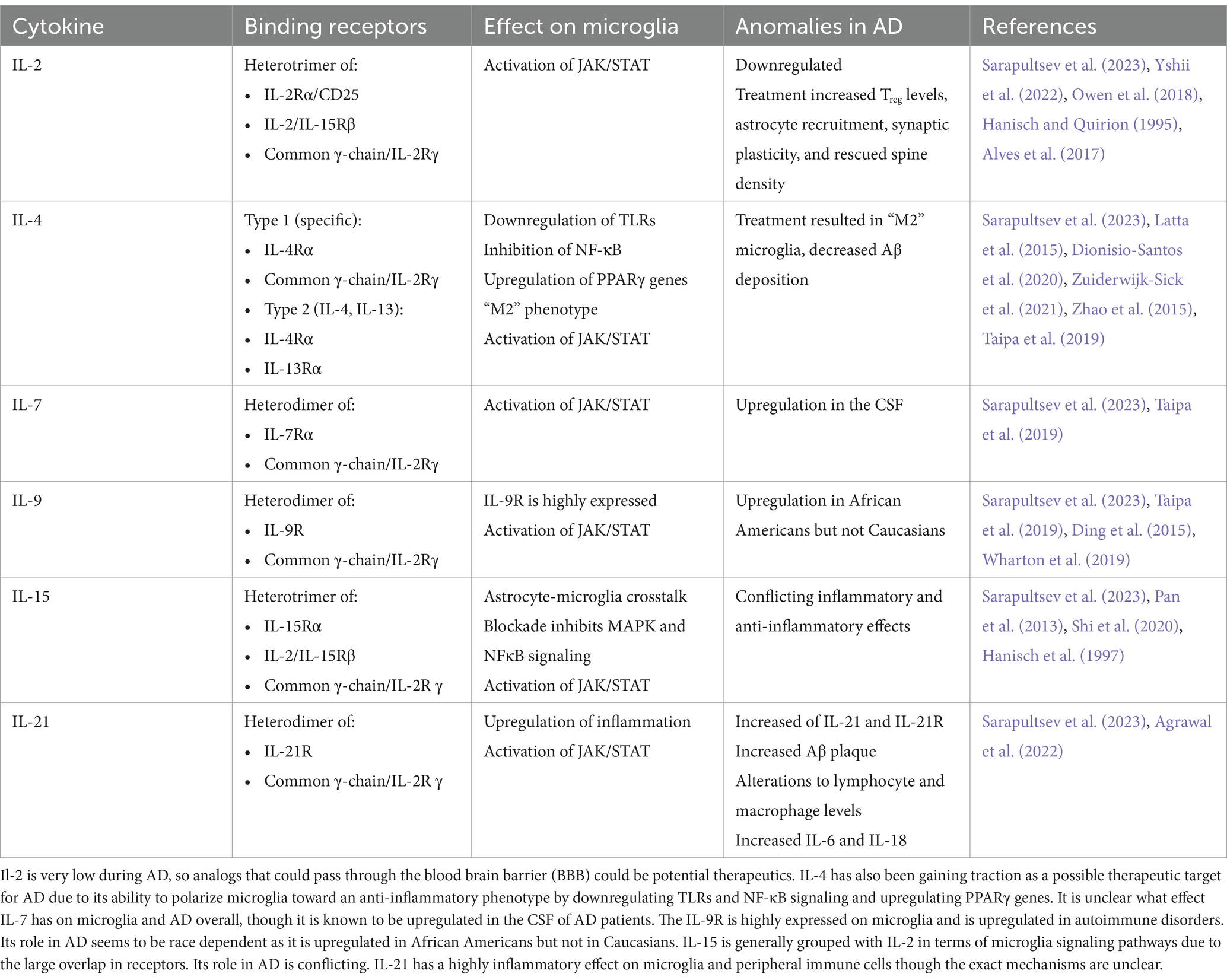
Table 1. Effect of common cytokine receptor γ chain cytokines on microglia and AD overall.
Modulation of these three signaling pathways to treat AD by reducing the highly inflammatory milieu of the brain has been gaining traction in the field. These pathways have been successfully targeted in other inflammatory disorders, so it is very appealing to repurpose relatively safe and inexpensive treatments for AD patients. The leading issue with these reutilizations is that the drugs often have low Blood–Brain Barrier (BBB) permeability. There are currently several clinical trials addressing the safety and efficacy of these inhibitors after promising results in pre-clinical studies.
2 MAPK signaling pathwayThe MAPK signaling pathway has three major components: the MAPK Kinase Kinase (MAPKKK), the MAPK Kinase (MAPKK), and MAPK. MAPKKK is usually phosphorylated by a protein kinase downstream of the cytokine receptor. MAPKKK phosphorylates MAPKK, which in turn phosphorylates MAPK. MAP Kinases are the third major kinase activated in the MAPK signaling pathway and are well-known for their roles in cellular proliferation, differentiation, or death. These kinases can be grouped into three families: ERK (extracellular-signal-regulated kinases), JNK (Jun amino-terminal kinases), and p38/SAPK (stress-activated protein kinases). We refer the reader to the excellent review by Morrison for detailed descriptions of the signaling mechanisms in these pathways (Morrison, 2012).
Depending on the specific kinases involved, MAPK signaling results in cell growth, survival, differentiation, inflammation, or apoptosis. Classic ERK signaling begins with a mitogen or growth factor and results primarily in cellular growth and differentiation (Morrison, 2012). JNK signaling is typically activated by environmental stress like oxidative stress, or inflammatory cytokines, but can also occur in response to growth factors, resulting in inflammation and metabolic changes (Morrison, 2012). It may also play a role in apoptosis, but whether it is apoptotic, anti-apoptotic, or uninvolved is unresolved (Morrison, 2012; Lin, 2003). P38 signaling, like JNK, is typically activated by environmental stress or inflammatory cytokines primarily resulting in inflammation, differentiation, apoptosis, or cell cycle regulation (Morrison, 2012). In AD, ERK, JNK, and p38 are upregulated in vulnerable neurons indicating involvement in AD pathogenesis.
Less is known about the role of MAPK in microglia. A recent study has indicated p-ERK expression is linked to microglia adopting a “DAM” phenotype and damaging inflammation in both an amyloid mouse model (5xFAD) and post-mortem patient samples (Chen et al., 2021). ERK signaling is activated downstream of the RTKs CSF1R and MERTK in homeostatic microglia, while it is activated by AXL and FLT1 in DAMs (Chen et al., 2021). It is critical in the regulation of IFNγ-mediated inflammation (Chen et al., 2021). Furthermore, ERK is upstream of several DAM and human AD risk genes such as TREM2, Tyrobp, Bin1, Cd33, Trem2, and Cnn2 (Chen et al., 2021). Analysis of post-mortem human brains found that ERK1 and ERK2 were the only MAPK signaling proteins with increased protein expression and positive associations with neuropathological grade (Chen et al., 2021).
JNK is mostly activated in AD by the environmental stress of the APP cleavage product amyloid-β (Aβ) (Solas et al., 2023). When Aβ was given to wild type mice, p-JNK levels were significantly elevated (Solas et al., 2023). In a follow-up experiment, JNK3 overexpression via a viral vector caused cognitive deficiencies and tau misfolding in Tg2576 mice but did not impact amyloid pathology (Solas et al., 2023). Taken together, this suggests that JNK signaling is in response to Aβ but affects tau pathology (Solas et al., 2023). Whether JNK activation precedes or is in response to amyloid plaque in AD patients is under debate. p-JNK was significantly increased in the post-mortem brains of AD patients, but it was also upregulated in non-AD related dementia patients (Solas et al., 2023). However, there was a co-localization between p-JNK and Aβ (Solas et al., 2023).
P38 is mainly expressed by brain resident immune cells (glia) during neurodegeneration though previous research has mostly focused on its role in neurons mostly due to its ability to phosphorylate tau and consequently worsen pathology (Perea et al., 2022; Asih et al., 2020). In a tau mouse model (P301S), p38 activation increased during aging primarily in hippocampal microglia (Perea et al., 2022). Interestingly, this model produced p38low microglia that seemed to have a neuroprotective effect in this context (Perea et al., 2022).
All three families of MAP kinases stimulate inflammatory cytokine production in microglia. Microglia treated with Aβ or APP showed activation of the three MAPKs (Kim et al., 2004; Bodles and Barger, 2005). Interestingly, only JNK and p38 inhibitors reduced both nitrite and nitric oxide synthase (iNOS) accumulation, indicating only JNK and p38 are involved in microglial activation that results in neuronal damage (Bodles and Barger, 2005). This has also been seen in a microglial-neuronal co-culture experiment where inhibitors to JNK and p38, but not ERK, prevented neuronal death (Xie et al., 2004). Preliminary experiments implicate Toll-Like Receptor 4 (TLR4) upregulation on microglia resulting in JNK and p38 mediated neuronal damage though there has not been follow-up (Gaikwad et al., 2015).
Two p38 inhibitors, MW150 and Neflamapimod, are currently in clinical trials for AD treatment. Both were developed to mitigate the off-target effects on the heart and liver often seen with first generation p38α inhibitors caused by non-p38 kinases being inhibited at higher concentrations (Libby and Everett, 2019; Hammaker and Firestein, 2010). MW150 is a p38α inhibitor developed by Neurokine Therapeutics that is currently in a phase 2a randomized double-blind, placebo-controlled, study, in mild-to-moderate AD patients though results have not yet been released. Fourteen-day treatment with MW150 improved symptoms across animal models though the specific effects differed. In one study, improved synaptic function and cognition was observed in both older and younger animals (Rutigliano et al., 2018). However, in another study, treatment did not affect plaque load or immune cells markers but did decrease inflammatory cytokine levels (Zhou et al., 2017). Another study found that MW150 improved hippocampal-dependent memory in two AD animal models while also being highly specific to p38α (Roy et al., 2015). It is hoped that the specificity and robustness of MW150 treatment in animal models will translate into the same response in patients.
Neflamapimod, developed by EIP Pharma, is also in phase 2 studies to test efficacy. In an open-label clinical study, patients had improved immediate and delayed recall compared to baseline, correlating with drug plasma levels (Scheltens et al., 2018). It was also able to cross the BBB and decrease TNF-α and IL-8 in the CSF. However, it should be noted that this study was not placebo controlled (Scheltens et al., 2018; Alam et al., 2017). It appears that Neflamapimod may be better suited to treating dementia caused by Lewy Bodies than dementia caused by AD, as it is currently in a Phase 2b (hypothesis-testing), multi-center, randomized, double-blind, placebo-controlled study for treating dementia with Lewy Bodies. It is possible that Neflamapimod could be revisited as a treatment for AD, especially if the current clinical trial is successful.
3 PI3K signaling pathwayThe PI3K pathway is most known for regulating cell survival though it also functions in cellular proliferation, growth, differentiation, and motility. PI3K is activated by kinases associated with the cytokine receptor. It then phosphorylates PIP2 to PIP3. PIP3 goes on to phosphorylate AKT, either directly or by activating mTORC2 or PDK1 that then activates AKT. AKT can act on numerous substrates but is most well-known for activating mTORC1, which is notably responsible for mRNA translation, autophagy, and mitochondrial functions (Sun et al., 2020). The pathway is generally inhibited by dephosphorylation of PIP3 into PIP2 by PTEN or dephosphorylation of AKT. The PI3K pathway in microglia is closely associated with many other signaling pathways, making it an attractive therapeutic target. Interestingly, PI3K and ERK signaling exhibit mutual inhibition (Manning and Toker, 2017).
While in a healthy brain, the PI3K pathway is associated with neuronal development and plasticity, in an AD brain, PI3K is associated with neuronal toxicity and inflammatory phenotypes of astrocytes and microglia. The PI3K/AKT pathway becomes dysregulated in AD, due to mutations that seemingly suppress its negative regulation (Chu et al., 2021; Tsai et al., 2021; Yoshino et al., 2017; Yao et al., 2019). Clinically, this represents a risk factor for late-onset AD and is linked to disease-related neuropathology (Chu et al., 2021; Efthymiou and Goate, 2017).
This pathway is generally accepted as the central regulator of microglial activation in response to stimulation. Most of this signaling is downstream of the TLR4 receptor, which is increased in AD (Chu et al., 2021; Fang et al., 2017). IL-4 can also activate PI3K signaling, but it is reduced in AD models (Gadani et al., 2012). Activation of PI3K signaling notably results in microglia secreting pro-inflammatory factors and can be further exacerbated by mitochondrial dysfunction increasing ROS and iNOS species that then activate NF-κB (Chu et al., 2021; Hoogland et al., 2018; Willis et al., 2020; Tönnies and Trushina, 2017; Nakajima and Kohsaka, 2001).
AKT activity may be neuroprotective in certain situations, especially in younger models as AKT exhibits age-related shifts in signaling (Razani et al., 2021). Microglia adopt a more chronic inflammatory phenotype that has reduced phagocytic capabilities during aging (Njie et al., 2012), though whether this results in or is an effect of AKT signaling is unclear. The role of mTORC proteins has also been inconsistent in AD mouse models. While the early use of mTORC inhibitors like rapamycin have decreased AD disease hallmarks, these inhibitors exacerbated damage in later disease stages, potentially due to the inhibition of mTORC1 interacting with TREM2, which improves amyloid plaque clearance (Razani et al., 2021; Shi et al., 2022). However, it is unclear whether this is a distinct signaling pathway from the classic inflammatory one.
Modulation of the PI3K pathway to treat AD has been gaining traction because it could affect both neuronal and glia cells. However, only rapamycin, an mTORC1 inhibitor, is currently in a phase 2 clinical trial. It should be noted that rapamycin is most effective at early disease stages and can have side effects (Razani et al., 2021; Majumder et al., 2011; Carosi and Sargeant, 2019). For those reasons, a second generation of mTORC inhibitors is being developed that can act on both mTORC1 and mTORC2, and more recently, dual mTORC and PI3K inhibitors have also been developed (Razani et al., 2021; Dowling et al., 2010; Benjamin et al., 2011; Zaytseva et al., 2012). One dual inhibitor, NVPBEZ235, was found to improve memory impairment and microglia activation in an AD mouse model (Razani et al., 2021; Bellozi et al., 2016; Bellozi et al., 2019).
4 JAK/STAT signaling pathwayThe JAK/STAT pathway is the backbone of many intracellular signaling processes due to rapid membrane-to-nucleus signaling. Unlike MAPK and PI3K signaling, for which the classical signaling occurs in the cytoplasm, JAK/STAT has a nuclear signaling component. The JAK kinases are non-covalently associated with receptors and are responsible for mediating tyrosine phosphorylation of receptors and recruiting STAT proteins. Tyrosine-phosphorylated STATs dimerize and are transported to the nucleus to regulate specific genes. JAK/STAT is seemingly the most prevalent of the three pathways in AD, as is responsible for the dysregulation of immune responses (Miao et al., 2023). It has been hypothesized that JAK inhibitors could be repurposed to help treat AD patients.
Mammals have four types of JAK and seven types of STAT, where the cytokine or growth factor initiating the cascade determines JAK and STAT protein activation (Table 2) (Sarapultsev et al., 2023). During neuroinflammation, these JAK/STAT signaling cascades typically result in microglia adopting an inflammatory phenotype. The notable exception to this is IL-4 activation of STAT6, mediating the re-polarization of microglia to an anti-inflammatory phenotype (Sarapultsev et al., 2023; He et al., 2020).
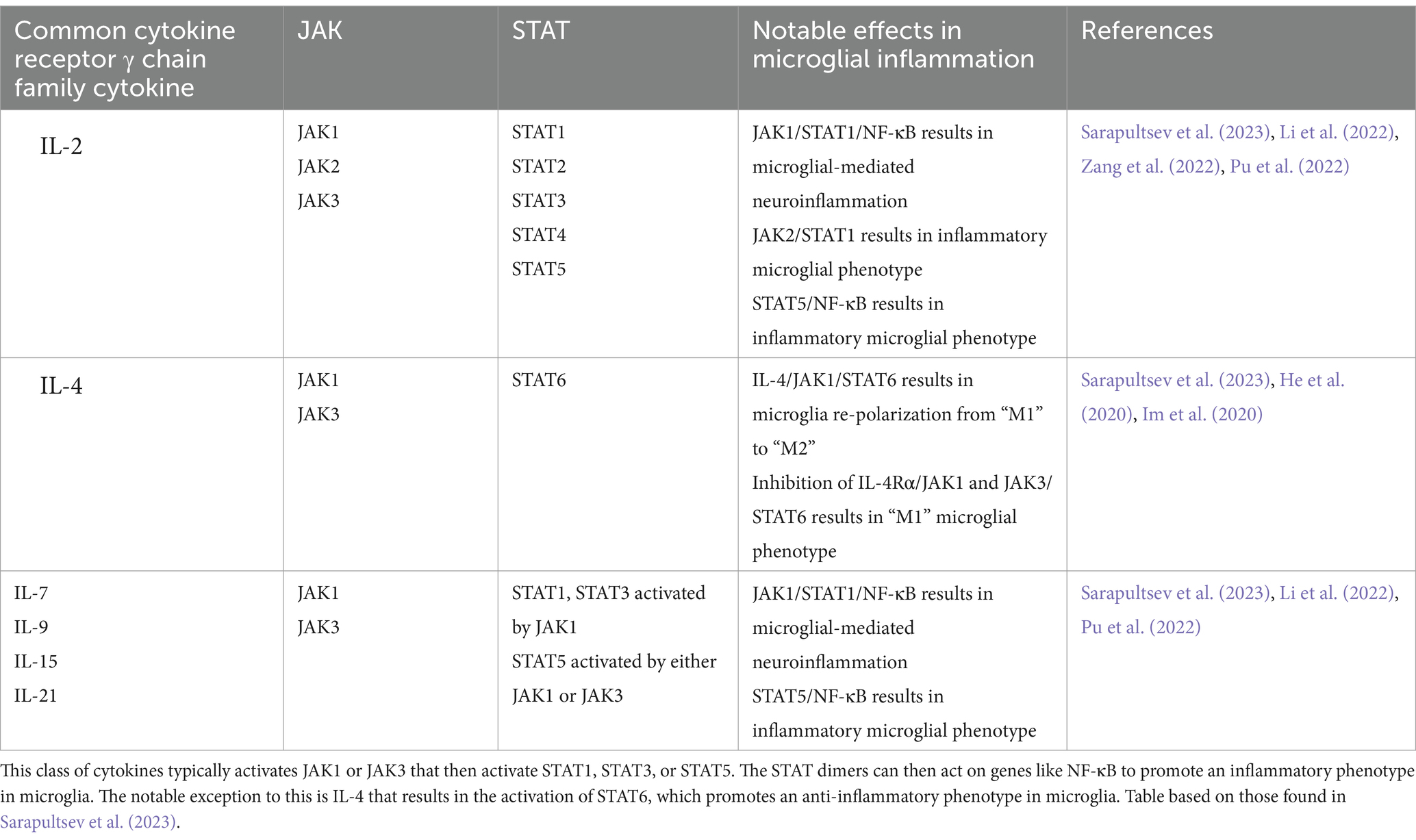
Table 2. JAK/STAT pathways activated by common cytokine receptor γ chain family cytokines and their notable effects in microglia.
Over-activation of the JAK/STAT pathway in AD is typically associated with STAT3, likely due to STAT3 phosphorylation being increased in the hippocampus of both mouse models and post-mortem brains (Millot et al., 2020). In addition, STAT3 may be a transcriptional regulator of BACE1, an important enzyme in Aβ production (Millot et al., 2020). STAT3 is known to be involved in microglia adopting a chronic inflammatory phenotype in response to Aβ and its inhibition significantly decreased microglia activation (Sarapultsev et al., 2023; Millot et al., 2020; Przanowski et al., 2014). This finding has been corroborated in an AD mouse model (APP/PS1) where STAT3 deficiency promoted Aβ phagocytosis by microglia (Doty et al., 2016). Interestingly, STAT3 signaling in response to Aβ shows time-dependent modifications. First, acetylated STAT3 levels increase, the consequence of which is currently unclear (Eufemi et al., 2015). This is followed by increased levels of phosphorylated STAT3, shifting the microglial proteome to one of chronic activation (Eufemi et al., 2015). Levels of 14–3-3ε, a known marker of Aβ-activated microglia, are eventually increased (Eufemi et al., 2015).
It has often been hypothesized that JAK/STAT inhibitors could be re-purposed to treat AD. Since these inhibitors are relatively safe and inexpensive, their re-utilization in AD to suppress neuroinflammation would greatly improve current treatment plans. However, low BBB permeability in most FDA-approved JAK/STAT inhibitors remains an issue. Hydroxychloroquine is a STAT3 inhibitor that is currently approved to treat rheumatoid arthritis and systemic lupus erythematosus. In rheumatoid arthritis patients, hydroxychloroquine treatment was associated with a lower risk of AD incident compared to methotrexate treatment, accounting for biases like informative censoring, reverse causality, and outcome misclassification (Varma et al., 2023). This was followed by analysis in an AD mouse model (APP/PS1) that found hydroxychloroquine is a potential treatment for AD by inactivating STAT3 in neurons, astrocytes, and microglia. This notably resulted in improved hippocampal synaptic plasticity, improved amyloid plaque clearance by microglia, decreased tau phosphorylation, and decreased neuroinflammation (Varma et al., 2023). It should be noted that these effects required treatment to start before significant plaque accumulation. JAK inhibitors baricitinib and tofacitinib were both evaluated as possible treatments for AD, but they were found to have a low likelihood of success due to low BBB permeability (Faquetti et al., 2024). Baricitinib, however, is currently in an open-label, biomarker-driven basket trial in patients with subjective cognitive disorder, mild cognitive impairment, Alzheimer’s Disease (AD), Amyotrophic lateral sclerosis (ALS), or asymptomatic carriers of an ALS-related gene at Massachusetts General Hospital.
5 ConclusionThe common cytokine receptor γ chain family of cytokines can activate three major signaling pathways in microglia to typically result in AD-promoting phenotypes (Figure 1). The MAPK pathway is associated with damaging inflammation, with all three MAPKs being notably altered in AD. The PI3K pathway is generally accepted as the central regulator of microglia activation in response to cytokines though there are conflicting results on the downstream effects of PI3K activation; it is unclear whether conflicting results are due to distinct signaling pathways being affected. The JAK/STAT pathway is responsible for quick signaling associated with pro-inflammatory responses. Common cytokine receptor γ chain family of cytokines typically activate JAK1 or JAK3 that then activate STAT1, STAT3, or STAT5.
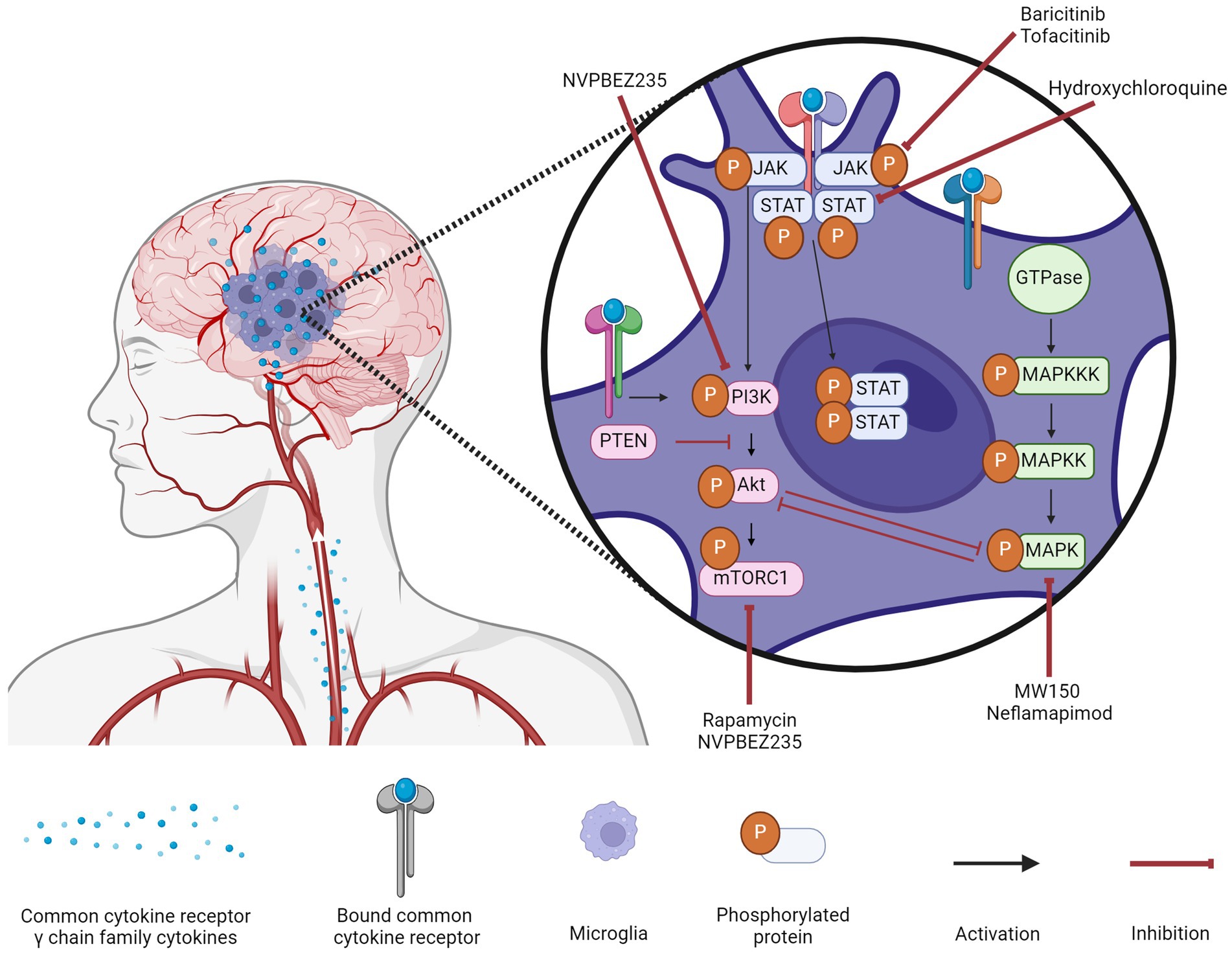
Figure 1. Microglial signaling cascades in Alzheimer’s disease and their inhibitors. Common cytokine receptor γ chain family cytokines activate three major signaling pathways (MAPK, PI3K, and JAK/STAT) in microglia during AD. A variety of inhibitors to components of these signaling cascades are being assessed for their efficacy in ameliorating the inflammatory milieu of the AD brain. Created with biorender.com.
The modulation of these three signaling pathways to treat AD (Figure 1) has been gaining traction though the clinical results are limited. Targeting the MAPK pathway can be difficult as upregulation of a specific kinase is not a guarantee that its inhibition will improve symptoms, as is the case with ERK kinases. Targeting the PI3K pathway could have the greatest effect given the numerous downstream targets, but for that same reason, it is also the most difficult to predict. While the backbone of the pathway is well-characterized, there is a lack of research into the downstream effects when different components of the core pathway are inhibited. Until these effects are identified, it will be difficult to transition from pre-clinical studies to clinical ones. Targeting the JAK/STAT pathway is popular due to FDA-approved JAK inhibitors being relatively safe and inexpensive, but low BBB permeability remains the greatest obstacle for this repurposing.
Data availability statementThe original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributionsHZ: Conceptualization, Writing – original draft, Writing – review & editing. ER: Funding acquisition, Supervision, Writing – review & editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health (1R01AG075897) and The BrightFocus Foundation (A2021036S) to ER.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ReferencesAgrawal, S., Baulch, J. E., Madan, S., Salah, S., Cheeks, S. N., Krattli, R. P., et al. (2022). Impact of IL-21-associated peripheral and brain crosstalk on the Alzheimer’s Disease neuropathology. Cell. Mol. Life Sci. 79:331. doi: 10.1007/s00018-022-04347-6
Crossref Full Text | Google Scholar
Alam, J., Blackburn, K., and Patrick, D. (2017). Neflamapimod: clinical phase 2b-ready Oral small molecule inhibitor of P38α to reverse synaptic dysfunction in early Alzheimer’s Disease. J. Prev Alzheimers Dis. 4, 273–278. doi: 10.14283/jpad.2017.41
Crossref Full Text | Google Scholar
Alves, S., Churlaud, G., Audrain, M., Michaelsen-Preusse, K., Fol, R., Souchet, B., et al. (2017). Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s Disease mice. Brain 140, 826–842. doi: 10.1093/brain/aww330
Crossref Full Text | Google Scholar
Asih, P. R., Prikas, E., Stefanoska, K., Tan, A. R. P., Ahel, H. I., and Ittner, A. (2020). Functions of P38 MAP kinases in the central nervous system. Front. Mol. Neurosci. 13:570586. doi: 10.3389/fnmol.2020.570586
PubMed Abstract | Crossref Full Text | Google Scholar
Bellozi, P. M. Q., Lima, I. V. D. A., Dória, J. G., Vieira, É. L. M., Campos, A. C., Candelario-Jalil, E., et al. (2016). Neuroprotective effects of the anticancer drug NVP-BEZ235 (Dactolisib) on amyloid-β 1-42 induced neurotoxicity and memory impairment. Sci. Rep. 6:25226. doi: 10.1038/srep25226
PubMed Abstract | Crossref Full Text | Google Scholar
Bellozi, P. M., Quaglio, G. F., Gomes, L. R., de Oliveira, I., Olmo, G., Vieira, É. L. M., et al. (2019). NVP-BEZ235 (Dactolisib) has protective effects in a transgenic mouse model of Alzheimer’s Disease. Front. Pharmacol. 10:1345. doi: 10.3389/fphar.2019.01345
PubMed Abstract | Crossref Full Text | Google Scholar
Benjamin, D., Colombi, M., Moroni, C., and Hall, M. N. (2011). Rapamycin passes the torch: a new generation of MTOR inhibitors. Nat. Rev. Drug Discov. 10, 868–880. doi: 10.1038/nrd3531
PubMed Abstract | Crossref Full Text | Google Scholar
Bodles, A. M., and Barger, S. W. (2005). Secreted β-amyloid precursor protein activates microglia via JNK and P38-MAPK. Neurobiol. Aging 26, 9–16. doi: 10.1016/j.neurobiolaging.2004.02.022
Crossref Full Text | Google Scholar
Carosi, J. M., and Sargeant, T. J. (2019). Rapamycin and Alzheimer Disease: a double-edged sword? Autophagy 15, 1460–1462. doi: 10.1080/15548627.2019.1615823
Crossref Full Text | Google Scholar
Chen, M. J., Ramesha, S., Weinstock, L. D., Gao, T., Ping, L., Xiao, H., et al. (2021). Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s Disease. J. Neurosci. Res. 99, 1704–1721. doi: 10.1002/jnr.24829
PubMed Abstract | Crossref Full Text | Google Scholar
Chu, E., Mychasiuk, R., Hibbs, M. L., and Semple, B. D. (2021). Dysregulated phosphoinositide 3-kinase signaling in microglia: shaping chronic Neuroinflammation. J. Neuroinflammation 18:276. doi: 10.1186/s12974-021-02325-6
Crossref Full Text | Google Scholar
Ding, X., Cao, F., Cui, L., Ciric, B., Zhang, G.-X., and Rostami, A. (2015). IL-9 signaling affects central nervous system resident cells during inflammatory stimuli. Exp. Mol. Pathol. 99, 570–574. doi: 10.1016/j.yexmp.2015.07.010
Crossref Full Text | Google Scholar
Dionisio-Santos, D. A., Behrouzi, A., Olschowka, J. A., and Kerry O’Banion, M. (2020). Evaluating the effect of Interleukin-4 in the 3xTg mouse model of Alzheimer’s Disease. Front. Neurosci. 14:441. doi: 10.3389/fnins.2020.00441
PubMed Abstract | Crossref Full Text | Google Scholar
Doty, K. R., Guillot-Sestier, M.-V., and Town, T. (2016). P4-304: STAT3 signaling referees microglial amyloid clearance in Alzheimer’S Disease. Alzheimers Dement. 12:P1150. doi: 10.1016/j.jalz.2016.07.047
Crossref Full Text | Google Scholar
Dowling, R. J. O., Topisirovic, I., Fonseca, B. D., and Sonenberg, N. (2010). Dissecting the role of MTOR: lessons from MTOR inhibitors. Biochim. Biophys. Acta. 1804, 433–439. doi: 10.1016/j.bbapap.2009.12.001
Crossref Full Text | Google Scholar
Efthymiou, A. G., and Goate, A. M. (2017). Late onset Alzheimer’s Disease genetics implicates microglial pathways in Disease risk. Mol. Neurodegener. 12:43. doi: 10.1186/s13024-017-0184-x
Crossref Full Text | Google Scholar
Eufemi, M., Cocchiola, R., Romaniello, D., Correani, V., Di Francesco, L., Fabrizi, C., et al. (2015). Acetylation and phosphorylation of STAT3 are involved in the responsiveness of microglia to Beta amyloid. Neurochem. Int. 81, 48–56. doi: 10.1016/j.neuint.2015.01.007
PubMed Abstract | Crossref Full Text | Google Scholar
Fang, W., Bi, D., Zheng, R., Cai, N., Hong, X., Zhou, R., et al. (2017). Identification and activation of TLR4-mediated Signalling pathways by alginate-derived Guluronate oligosaccharide in RAW264.7 macrophages. Sci. Rep. 7:1663. doi: 10.1038/s41598-017-01868-0
PubMed Abstract | Crossref Full Text | Google Scholar
Faquetti, M. L., Slappendel, L., Bigonne, H., Grisoni, F., Schneider, P., Aichinger, G., et al. (2024). Baricitinib and tofacitinib off-target profile, with a focus on Alzheimer’s disease. Alzheimers Dement (NY). 10:e12445. doi: 10.1002/trc2.12445
Crossref Full Text | Google Scholar
Gaikwad, S., Patel, D., Naveen, C., and Agrawal-Rajput, R. (2015). The critical role of JNK and P38 MAPKs for TLR4 induced microglia-mediated neurotoxicity. Eur. J. Exp. Biol. 5, 34–42. doi: 10.1155/2015/361326
Crossref Full Text | Google Scholar
Hammaker, D., and Firestein, G. S. (2010). ‘Go upstream, young man’: lessons learned from the P38 Saga. Ann. Rheum. Dis. 69, i77–i82. doi: 10.1136/ard.2009.119479
Crossref Full Text | Google Scholar
Hanisch, U. K., and Quirion, R. (1995). Interleukin-2 as a Neuroregulatory cytokine. Brain Res. Brain Res. Rev. 21, 246–284. doi: 10.1016/0165-0173(95)00015-1
Crossref Full Text | Google Scholar
Hanisch, U.-K., Lyons, S. A., Prinz, M., Nolte, C., Weber, J. R., Kettenmann, H., et al. (1997). Mouse brain microglia express Interleukin-15 and its Multimeric receptor complex functionally coupled to Janus kinase activity. J. Biol. Chem. 272, 28853–28860. doi: 10.1074/jbc.272.46.28853
Crossref Full Text | Google Scholar
He, Y., Gao, Y., Zhang, Q., Zhou, G., Cao, F., and Yao, S. (2020). IL-4 switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience 437, 161–171. doi: 10.1016/j.neuroscience.2020.03.008
PubMed Abstract | Crossref Full Text | Google Scholar
Hoogland, I. C. M., Westhoff, D., Engelen-Lee, J.-Y., Melief, J., Serón, M. V., Houben-Weerts, J. H. M. P., et al. (2018). Microglial activation after systemic stimulation with lipopolysaccharide and Escherichia Coli. Front. Cell. Neurosci. 12:110. doi: 10.3389/fncel.2018.00110
PubMed Abstract | Crossref Full Text | Google Scholar
Im, J. H., Yeo, I. J., Park, P. H., Choi, D. Y., Han, S.-B., Yun, J., et al. (2020). Deletion of Chitinase-3-like 1 accelerates stroke development through enhancement of Neuroinflammation by STAT6-dependent M2 microglial inactivation in Chitinase-3-like 1 knockout mice. Exp. Neurol. 323:113082. doi: 10.1016/j.expneurol.2019.113082
Crossref Full Text | Google Scholar
Kim, S. H., Smith, C. J., and Van Eldik, L. J. (2004). Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1 Beta production. Neurobiol. Aging 25, 431–439. doi: 10.1016/S0197-4580(03)00126-X
Crossref Full Text | Google Scholar
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s Disease. Alzheimers Dement. 4, 575–590. doi: 10.1016/j.trci.2018.06.014
PubMed Abstract | Crossref Full Text | Google Scholar
Latta, C. H., Sudduth, T. L., Weekman, E. M., Brothers, H. M., Abner, E. L., Popa, G. J., et al. (2015). Determining the role of IL-4 induced Neuroinflammation in microglial activity and amyloid-β using BV2 microglial cells and APP/PS1 transgenic mice. J. Neuroinflammation 12:41. doi: 10.1186/s12974-015-0243-6
PubMed Abstract | Crossref Full Text | Google Scholar
Li, T., Li, L., Peng, R., Hao, H., Zhang, H., Gao, Y., et al. (2022). Abrocitinib attenuates microglia-mediated Neuroinflammation after traumatic brain injury via inhibiting the JAK1/STAT1/NF-ΚB pathway. Cells 11:3588. doi: 10.3390/cells11223588
PubMed Abstract | Crossref Full Text | Google Scholar
Libby, P., and Everett, B. M. (2019). Novel Antiatherosclerotic therapies. Arterioscler. Thromb. Vasc. Biol. 39, 538–545. doi: 10.1161/ATVBAHA.118.310958
Crossref Full Text | Google Scholar
Lin, A. (2003). Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25, 17–24. doi: 10.1002/bies.10204
Crossref Full Text | Google Scholar
Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2011). Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 6:e25416. doi: 10.1371/journal.pone.0025416
Crossref Full Text | Google Scholar
Manning, B. D., and Toker, A. (2017). AKT/PKB signaling: navigating the network. Cell 169, 381–405. doi: 10.1016/j.cell.2017.04.001
Crossref Full Text | Google Scholar
Miao, J., Ma, H., Yang, Y., Liao, Y., Lin, C., Zheng, J., et al. (2023). Microglia in Alzheimer’s Disease: pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 15:1201982. doi: 10.3389/fnagi.2023.1201982
PubMed Abstract | Crossref Full Text | Google Scholar
Millot, P., San, C., Bennana, E., Hugon, J., Paquet, C., Hosten, B., et al. (2020). STAT3 inhibition reverses Neuroinflammation and Aβ metabolism induced by systemic inflammation. Alzheimers Dement. 16:e041019. doi: 10.1002/alz.041019
Crossref Full Text | Google Scholar
Nakajima, K., and Kohsaka, S. (2001). Microglia: activation and their significance in the central nervous system. J. Biochem. 130, 169–175. doi: 10.1093/oxfordjournals.jbchem.a002969
Crossref Full Text | Google Scholar
Njie, E. G., Boelen, E., Stassen, F. R., Steinbusch, H. W. M., Borchelt, D. R., and Streit, W. J. (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 33, 195.e1–195.e12. doi: 10.1016/j.neurobiolaging.2010.05.008
Crossref Full Text | Google Scholar
O’Connor, J. L., and Nissen, J. C. (2023). The pathological activation of microglia is modulated by sexually dimorphic pathways. Int. J. Mol. Sci. 24:4739. doi: 10.3390/ijms24054739
PubMed Abstract | Crossref Full Text | Google Scholar
Ocañas, S. R., Ansere, V. A., Kellogg, C. M., Isola, J. V. V., Chucair-Elliott, A. J., and Freeman, W. M. (2023). Chromosomal and gonadal factors regulate microglial sex effects in the aging brain. Brain Res. Bull. 195, 157–171. doi: 10.1016/j.brainresbull.2023.02.008
留言 (0)