
記住我







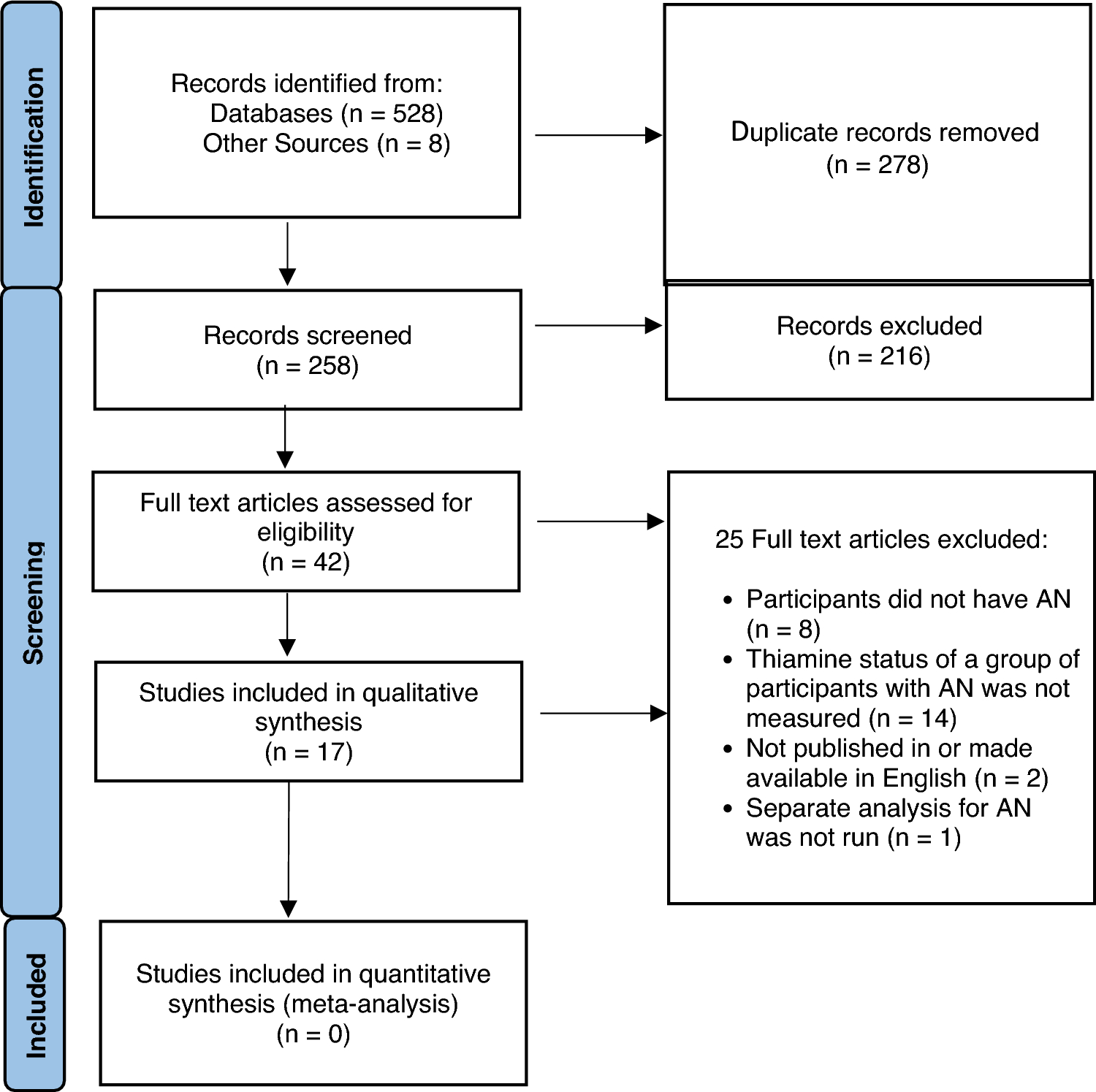

In the 6-month extension study, 662 patients were enrolled (APTS), of whom 79.5% (n = 526) completed the study, and 98.6% (n = 653) were included in the FAS for efficacy assessments (Fig. 2). Overall, 20.5% (n = 136) of the patients withdrew from the 6-month study. The most common primary reasons for withdrawal (> 4%) were “other” (5.9%, n = 39) and “adverse event” (5.1%, n = 34); six patients in the “other” category were withdrawn due to study termination based on the negative outcome of the short-term, double-blind, lead-in studies. In the 18-month extension study, 94 patients who completed the 6-month extension study were enrolled after which enrollment was stopped as requirement for sample size was met. All 94 were treated (APTS), of whom 61.7% (n = 58) completed treatment, and 94.6% (n = 89) were included in the FAS for efficacy assessments. A total of 38.3% (n = 36) of patients withdrew from the study. The most common primary reasons for withdrawal (> 4%) were “other” (28.7%, n = 27) and “withdrawal of consent” (4.3%, n = 4). Reasons for withdrawal marked as “other” included sponsor’s decision to withdraw adolescent patients based on the negative outcome of the lead-in adolescent study (n = 10), withdrawal of consent (n = 4), refusal to attend (n = 4), relocation or logistics (n = 3), not requiring medication anymore (n = 3), and 1 patient each due to non-adherence (parental concern), disallowed concomitant medication, or were in remission for a year. No patients withdrew due to an AE during the 18-month extension study.
Fig. 2
Patient disposition. AEs = adverse events; APTS = all-patients-treated set; FAS = full analysis set; n = number of patients
Demographic and baseline characteristicsThe demographic characteristics of the patients are provided in Table 1. In the 6-month extension study, 45.3% of patients were children (7–11 years) and 54.7% were adolescents (12–17), whereas in the 18-month extension study, 22.3% were children and 77.7% were adolescents. Approximately half the children were prepubertal (Tanner stage I), while among adolescents, a majority were in pubertal stages (Tanner stages II to IV) at the 6- and 18-month baselines. At the 6-month study baseline, the mean CDRS-R total score was 44.45, which further decreased to 33.42 at the 18-month study baseline.
Table 1 Patient demographicsSafety and tolerability outcomesAdverse eventsIn the 6-month extension study, 61% (n = 404) of patients reported treatment-emergent AEs (TEAEs), with the majority being mild to moderate, and 3.9% (n = 26) of patients experiencing a severe TEAE. As per the treatment, during the lead-in, double-blind, child and adolescent studies, the incidence of TEAEs in the 6-month extension study was similar among patients who received placebo (61.5%), vortioxetine 10 mg/day (58.0%), vortioxetine 20 mg/day (64.1%), and fluoxetine 20 mg/day (60.0%); nausea was reported in 26.0%, 19.5%, 19.6%, and 17.8%, for patients who received placebo, vortioxetine 10 mg/day, vortioxetine 20 mg/day, and fluoxetine 20 mg/day, respectively, in the lead-in studies. Overall, approximately 37% (n = 242) of patients experienced TEAEs that were considered causally related to the treatment. A total of 6.0% (n = 40) of patients withdrew because of a TEAE in the 6-month extension study, with nausea (1.7%; n = 11) being the most common TEAE leading to withdrawal, followed by suicidal ideation and suicide attempt (n = 4; 0.6% each). A total of 2.1% (n = 14) of patients reported serious AEs (SAEs), of which events related to psychiatric disorders were most common and were observed in 1.4% (n = 9). The SAEs that were reported in > 1 patient in the treatment period were suicide attempt (4 patients, 0.6%), suicidal ideation (4 patients, 0.6%), and intentional overdose (3 patients, 0.5%). TEAEs with an incidence of ≥ 5% in the 6-month study included nausea, headache, vomiting, nasopharyngitis, and abdominal pain (Table 2).
During the 18-month extension study, 51% (n = 48) of patients reported TEAEs, the majority of which were mild to moderate, with only 1 patient experiencing a severe TEAE. Approximately 18% (n = 17) of patients experienced TEAEs that were considered causally related to treatment. In the 18-month extension study, no patients withdrew because of a TEAE, and no SAEs or fatal events were reported. TEAEs with an incidence ≥ 5% in the 18-month extension study included nausea, headache, vomiting, nasopharyngitis, and abdominal pain (Table 2).
Table 2 TEAEs with ≥ 5% incidenceThere was a general decrease over time in the incidence and intensity of the signs and symptoms collected using the PAERS in both studies. The most common symptoms appeared to be related to MDD (mood-related symptoms, anxiety) and the comorbid disorder of ADHD (hyperactivity, attention difficulties). The symptoms also reflected some of the AEs in these studies, such as somnolence, stomachache, and nausea. In both studies, the majority of the PAERS symptoms were either mild or moderate.
Suicide-related eventsResults from the C-SSRS assessment in the 6-month extension study showed that 94% (617/659) of patients had no suicidal ideation or behavior during the study; five patients (0.8%) had suicidal behavior reported as SAEs (nonfatal suicide attempt, n = 4; suicidal behavior, n = 1), five patients (0.8%) had nonspecific active suicidal thoughts, and five patients (0.8%) had active suicidal ideation without intent to act. Suicidal ideation and suicide attempt were each reported by four patients (0.6%) as TEAEs leading to withdrawal from the 6-month extension period.
As per the C-SSRS assessment in the 18-month extension study, 96% (90/94) of patients had no suicidal ideation or behavior; 4.3% (n = 4) had suicidal ideation without intent to act, and no patients had suicidal behavior. One patient had a suicide-related TEAE captured by standardized MedDRA suicide/self-injury queries that was considered mild and not related to treatment.
Laboratory and vital signs findingsIn the 6-month extension study, mean changes from baseline to 6 months were small, and no consistent trends were observed for clinical safety laboratory tests, height, weight, BMI, vital signs, or ECG. Similarly, in the 18-month extension study, mean changes from baseline to 18 months were small, and no consistent trends were observed for clinical safety laboratory parameters, vital signs, height, weight, or BMI, except for prolactin, which showed an increase in mean score in five patients. At the end of the treatment period, these levels decreased to the baseline values for three patients, and the other two patients were not tested after the end of study. In addition, one patient showed a shift from normal weight to obese during the 18-month extension study. In both the 6- and 18-month studies, there were no notable changes observed in reproductive hormones, Tanner score, or menstrual cycle duration from respective baselines to either the 6-month or 18-month time points.
Effectiveness outcomesIn the 6-month extension study, the change in mean CDRS-R total score and mean CGI-S from baseline to 6 months was − 16.7 and − 1.5 points, respectively, showing improvement in MDD symptom severity (Supplementary Table). Likewise, the CGI-I score was 1.7 points at 6 months, suggesting improvement. At 6 months, more than 85% of patients demonstrated a response (CGI-I score ≤ 2) and 59% were in remission (CDRS-R total score ≤ 28) (Table 3). Remission rates were similar for patients from both short-term, double-blind, lead-in studies, with 58% of patients from the child study and 60% from the adolescent study in remission at 6 months. Cognitive improvement was demonstrated through reduction in BRIEF-P and BRIEF-SR scores from baseline to 6 months by > 7 points. The CGAS scores increased by more than 14 points from baseline to 6 months, indicating improvement in overall functioning. Confirming these assessments, patients also reported improvement in PedsQL™ VAS total and emotional distress scores.
In the 18-month extension study, the change in mean CDRS-R total score and mean CGI-S from the baseline to 18 months was − 8.9 and − 1.3 points, respectively, showing further improvement in MDD symptoms from the end of the 6-month study (Table 3). The CGI-I score was 1.4 points at 18 months, also showing improvement. A total of 84% of patients were in remission (CDRS-R total score ≤ 28) at 18 months (Table 3); 87% of patients from the double-blind child lead-in study and 82% from the adolescent study were in remission at 18 months. At 18 months, the reduction in BRIEF-P scores was > 7 points and the reduction in BRIEF-SR score was > 11 points from baseline. The CGAS scores increased by more than 11 points from baseline to 18 months, indicating good overall functioning, and similar improvements were noted through the PedsQL™ VAS total and emotional distress scores.
Table 3 Response and remission analysisTreatment course and adherenceDuring the 6-month extension study, the mean total duration of exposure based on tablet count was approximately 160 days (~ 5 months) and was similar in patients from both the child and adolescent double-blind, lead-in studies. The mean dose of vortioxetine was 13 mg/day, with patients from the child lead-in study generally receiving a slightly lower mean dose than those from the adolescent study (12 mg/day vs. 13 mg/day). Most patients (≥ 97%) demonstrated ≥ 80% adherence to the treatment based on the tablet count; however, 110 (25%) of the 434 patients treated with vortioxetine demonstrated nonadherence based on plasma concentrations in terms of estimated apparent total plasma clearance of drug values (estimated CL/F > 120 L/h) and PK samples below the first lower limit of quantification at any visit when it was assessed. During the 18-month extension study, approximately 74% of patients received vortioxetine for at least 15 months; a majority of patients (98.9%) received the intended dose of vortioxetine and demonstrated ≥ 80% adherence to treatment based on tablet count. The mean dose of vortioxetine was 13 mg/day, with patients from the child lead-in study generally receiving a lower mean dose than those from the adolescent study (11 mg/day vs. 14 mg/day). The modal dose of vortioxetine was 10 mg for both the 6- and 18-month extension studies, with similar mean doses of 12.4 mg at 6 months and 13.2 mg at 18 months (Table 4).
Table 4 Exposure to vortioxetine at 6 and 18 months
留言 (0)