
記住我





STS played a pivotal role as the first model illustrating the potential of immunotherapy in cancer treatment, tracing back to the 19th century when William Coley observed sarcoma regression following bacterial infection clearance [35]. In the present landscape of immuno-oncology, significant strides have been made, with approvals for administration granted in various solid tumors, including melanoma, non-small cell lung cancer, and renal cell carcinoma, among others. However, despite this progress, the application of immunotherapy in STS remains a complex challenge. An analysis of combined data from nine clinical trials exploring immune checkpoint inhibitors (ICIs) in sarcomas involving 384 patients revealed an overall objective response rate (ORR) of 15.1% [36]. Nevertheless, upon excluding alveolar soft-part sarcoma, a rare subtype known for its heightened responsiveness to PD-1/PD-L1 monoclonal antibodies [37], the ORR diminished to 9.8% [36]. These disappointing results are mainly due to the high number of subtypes, some of which are ultrarare [37] and scattered information available about the tumor microenvironment (TME) for the various subtypes of sarcomas [38].
In an exhaustive analysis of the STS microenvironment, Petitprez et al. [39] introduced an immune classification of STS grounded in transcriptomic data, delineating five distinct sarcoma immune classes (SIC) characterized by varying levels of immune infiltration. The identification of tertiary lymphoid structures (TLS) emerged as a defining feature of the immune-high class, correlating with enhanced outcomes, and serving as a predictor for responses to immunotherapy. TLS are ectopic lymphoid formations that develop within non-lymphoid tissues, mirroring the structural and functional characteristics of lymph nodes [40]. These structures contain B-cell follicles and germinal centers surrounded by a T-cell region. In the context of antitumor immunity, TLS play a crucial role by fostering interactions between immune cells, promoting the activation and maturation of B and T cells, and enhancing local immune responses [40]. In the first biomarker-driven immunotherapy trial conducted in patients with STS, the existence of TLS has been correlated with improved outcomes and heightened responsiveness to immune-checkpoint inhibition. Notably, patients with TLS-positive STS exhibited an objective response rate of 30%, comparable to the rates observed in approved indications such as lung cancer and melanomas [41] (Fig. 1).
Fig. 1
A Representative example of tertiary lymphoid structure in a case of epithelioid sarcoma. B Locally advanced epithelioid sarcoma of the groin refractory to standard therapies. C Objective response after two infusions of pembrolizumab
These promising findings underscore the potential of immunotherapy as a more effective treatment option for certain sarcoma patients when compared to traditional cytotoxic chemotherapy. However, several pivotal questions remain. First, there is a need to explore strategies to further improve response rates to ICIs, specifically in sarcomas characterized by the presence of TLS. Additionally, addressing the challenge of achieving success with immunotherapy in the substantial 80% of patients who exhibit TLS-negative sarcoma is crucial for expanding the applicability of these therapeutic approaches. Last, a critical inquiry emerges regarding the generalizability of these findings to pediatric and adolescent and young adult cases of soft tissue and bone sarcomas, necessitating exploration of the potential benefits across diverse age groups within the sarcoma patient population.
Two ongoing randomized studies are currently exploring novel combinations to further enhance the response rate to PD-1 inhibition in patients with TLS-positive sarcomas. The CONGRATS study [NCT04095208] is investigating the combination of nivolumab with the LAG-3–blocking antibody relatlimab versus nivolumab alone in patients with advanced TLS-positive advanced or metastatic STS. Notably, LAG-3 in sarcomas is significantly upregulated in TLS-positive STS [39], and its expression has been associated with a poor outcome in comparison on transcriptomic analysis of over 600 complex genomics STS [42]. Recruitment for this study has recently concluded, and the results are anticipated in 2024.
Simultaneously, the TRUST study [NCT04874311] is exploring the combination of doxorubicin with a bifunctional fusion protein targeting TGF-β and PD-L1, bintrafusp alfa. Certain cytotoxic drugs, such as anthracyclines, the standard first-line treatment for advanced STS, can induce specific cellular responses beyond the typical apoptotic pathway, rendering tumor cell death immunogenic. Recent preclinical data from various tumor models demonstrated that appropriately selected immunogenic drugs, including anthracyclines, could sensitize tumors lacking T cell infiltration to host antitumor T cell immunity [43]. Moreover, instigating tumor infiltration by T cells sensitized tumors to checkpoint inhibition and durably controlled cancer. All these findings suggest that combining checkpoint blockade with immunogenic cytotoxic drugs can significantly expand the proportion of cancers responding to checkpoint therapy. Given that TGFB1 is highly overexpressed in TLS-positive sarcomas [39], the “proof of concept” in the TRUST study aims to prospectively demonstrate the high clinical benefit rate of PD-1/TGFB1 inhibition combined with doxorubicin versus doxorubicin alone in TLS-positive sarcomas. This study represents the first biomarker driven randomized investigation of an immunotherapy regimen in the first line setting for patients with advanced STS, and its results are eagerly anticipated in 2025.
For cold sarcomas, which constitute most cases, diverse therapeutic strategies are being explored to convert them into a “hot” and more responsive state to immune checkpoint inhibition. Notably, and as indicated above, immunogenic cytotoxic drugs such as anthracyclines play a crucial role, inducing specific cellular responses beyond traditional apoptotic pathways and rendering tumor cell death immunogenic. Wilky et al. conducted a phase 2 study combining doxorubicin with zalifrelimab (CTLA-4 inhibitor) and balstilimab (PD-1 inhibitor) in advanced STS [8]. The study aimed to improve 6-months PFS compared to historical doxorubicin. Among thirty enrolled patients with various STS types, four (12%) experienced grade 3/4 immune-related adverse events, including colitis, pancreatitis, diabetic ketoacidosis, hypertriglyceridemia, and hypothyroidism. Despite acceptable safety, the 6-month non-progression rate (NPR) was 46.4% (95% CI 28–66), falling short of the study’s objective of 63%. Martin-Broto et al. reported the efficacy and safety data of a combination of nivolumab with the doxorubicin/dacarbazine chemotherapy regimen [9]. Like the rationale reported by Wilky et al., this study included thirty-six patients with advanced leiomyosarcomas. Safety was acceptable, with 15% of patients experiencing grade 3/4 neutropenia. Nine patients achieved an objective response, six had stable disease, and one had progressive disease. The mPFS was 8.7 months (95% CI 7.9–9.3).
Beyond cytotoxic drugs, radiation therapy has also shown promise in impacting the tumor microenvironment and enhancing immune responses by releasing tumor antigens. In a limited series of 11 patients with STS, neoadjuvant radiation therapy led to a substantial rise in the overall immune cell infiltration within tumors across various histologic subtypes [44]. A significant elevation was noted in the proportion of monocytes and macrophages, specifically M2 macrophages, along with an increased presence of B cells and CD4 + T cells. Several studies investigating radiotherapy-immunotherapy regimens are currently underway in advanced STS and summarized in Table 3.
Table 3 Clinical trial combining immunotherapy and radiation therapyOncolytic viruses represent another potential approach that has shown significant immune-stimulating potential in preclinical settings. Hatta et al. demonstrated the potential of oncolytic viral therapy, specifically utilizing the third generation of HSV T-01, as a promising alternative to chemotherapy for refractory sarcomas [45]. In vitro and in vivo experiments in this study revealed the significant cytotoxic effects and replication capacity of T-01 in both rhabdomyosarcomas (RMS) and leiomyosarcomas (LMS). T-01 effectively suppressed tumor growth in subcutaneous tumor models of LMS and RMS, highlighting its immune-stimulating effects and potential as a novel therapeutic approach for cold sarcomas. The METROMAJX study investigated the systemic impact of JX-594, an oncolytic virus, in 15 patients with advanced STS, revealing an upregulation of antitumor immune response-related cytokines [46]. Although the clinical activity was low, the results of this study pave the way for innovative approaches to be evaluated in patients with advanced STS. The combination of oncolytic virus with immune checkpoint inhibition may represent one of them, as illustrated by the reported results of a single-center, phase II trial that investigated the combination of talimogene laherparepvec (T-VEC), an oncolytic immunotherapy derived from a modified human herpes simplex virus type 1, with pembrolizumab in patients with advanced STS [47]. T-VEC was administered intra-tumorally, with 20 patients enrolled in the trial. Most of them had locally advanced disease, and objective responses were observed in 30% of patients across five different histological subtypes. In the second part of the METROMAJX study, the response rate was lower. However, sequential biopsies revealed that intra-tumoral injection of JX-594 was capable of reshaping the microenvironment in TLS-negative sarcomas, thereby sensitizing them to PDL1 inhibition [48]. Several other studies investigating oncolytic viral therapies are currently underway and summarized in Table 4. Antiangiogenic therapy, by targeting tumor vasculature, may also reshape the microenvironment to enhance immune cell infiltration. Van Tine et al. recently reported the results of a randomized phase 2 study [11] investigating the activity of the VEGFR inhibitor cabozantinib combined with nivolumab and ipilimumab in a randomized phase 2 study versus cabozantinib alone. Sixty-nine patients were randomized to the combination arm, and 36 received cabozantinib monotherapy. The combination arm observed seven objective responses (11%), while the monotherapy arm had two (6%). The mPFS was 5.3 months (95% CI 4.1–11) for the combination and 3.5 months (95% CI 1.1–7.7) for monotherapy (p = 0.016). The mOS was 22.6 months (95% CI 14.8-NA) for the combination and not reached (95% CI 9.6-NA) for monotherapy (p = 0.42). Notably, among the 19 patients from the cabozantinib monotherapy arm who were allowed to crossover to the combination arm, seven showed tumor shrinkage, suggesting at least an additive effect of the combination of nivolumab and ipilimumab. These strategies collectively represent a multifaceted approach to enhancing tumor immunogenicity and improving responsiveness to immune checkpoint inhibitors. However, it is important to note that the majority of the recently reported or ongoing studies are single-arm and/or did not include analysis of sequential blood or tissue samples. Unfortunately, the absence of sequential tumor biopsies and randomization hinders drawing definitive conclusions regarding the influence of these combinations on the tumor microenvironment and their potential correlation with clinical benefits. Therefore, a paradigm shift in the design of immune-oncology trials in patients with STS is essential to enhance their scientific value and contribute to advancing knowledge in the field.
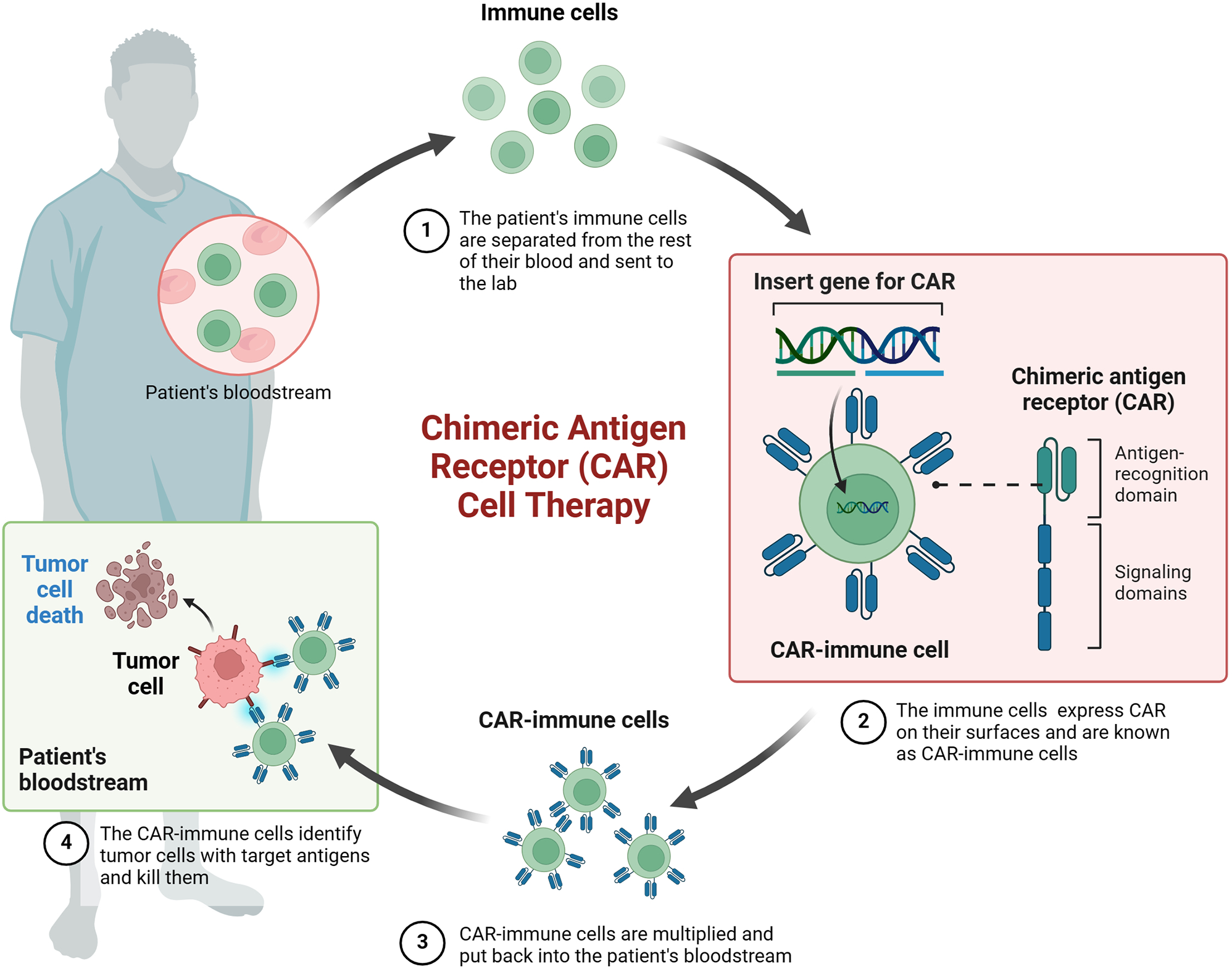
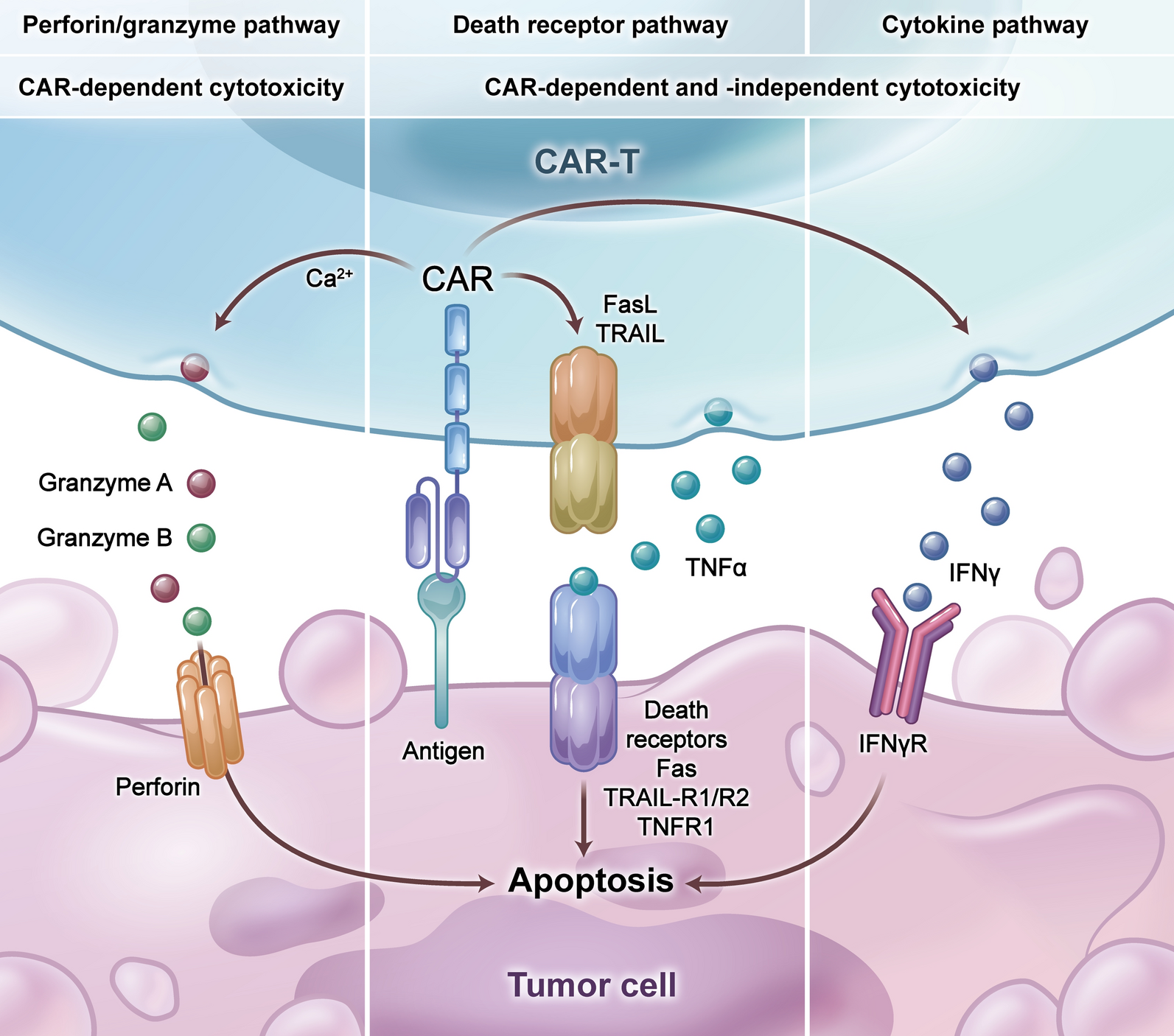
Table 4 Clinical trial testing oncolytic viral therapiesT cells are pivotal players in cell-mediated immunity, and the landscape of cancer treatment has witnessed notable advancements with the introduction of adoptive cell transfer (ACT) strategies, offering an alternative avenue in immunotherapy alongside immune checkpoint inhibition. Two prominent approaches in this realm are chimeric antigen receptor (CAR) T cell therapy and T cell receptor (TCR) T cell therapy, each unlocking new possibilities in the battle against malignant tumors.
In the spotlight is afami-cel, an autologous TCR T cell therapy specifically designed for HLA A*02–eligible patients with advanced solid tumors expressing the cancer testis antigen MAGE-A4. The ongoing SPEARHEAD-1 trial [NCT04044768] is actively assessing the efficacy and safety of afami-cel in individuals with advanced/metastatic synovial sarcomas (SS) or myxoid/round cell liposarcomas (LPS) [49].
The intricacies of this therapy unfold as autologous T cells are meticulously isolated and genetically engineered with a vector to express an affinity enhanced TCR, also known as a specific peptide enhanced affinity receptor T cell or SPEAR T cell. These modified T cells are primed to recognize and obliterate tumoral cells expressing the specific antigen MAGE-A4. Following this intricate process, the genetically enhanced T cells are expanded and reintroduced into the patient post-lymphodepletive chemotherapy.
Interim OS data from advanced SS patients treated with afami-cel paint an encouraging picture. The mOS reached 15.4 months (95% CI 10.9-NA), with 52% of patients censored at the data cutoff. The 12-month OS probability stood at 60%, while the 24-month OS probability reached 40%. Particularly noteworthy were the outcomes for the 17 patients with a RECIST response by independent review, where the median OS was not reached, and the 12-month and 24-month OS probabilities soared to 90% and 60%, respectively [50].
In the dynamic landscape of cancer therapeutics, afami-cel targeted approach, harnessing the power of genetically modified T cells against the specific antigen MAGE-A4, underscores the potential of ACT in addressing the complexities of advanced solid tumors such as SS and myxoid/round cell LPS. These results not only shed light on the promise of precision oncology but also pave the way for future advancements in personalized cancer treatments.
Targeted therapiesIn navigating the complex landscape of sarcoma treatment, the emergence of targeted therapies offers a beacon of hope, especially for select subtypes. This precision-oriented approach tailors treatments to the unique molecular profiles of these cancers, promising improved outcomes. As we explore the transformative potential of targeted therapies in STS management, it’s essential to acknowledge their selective impact, marking a new era of personalized care for specific subgroups within the sarcoma spectrum. While some strategies have been substantiated by compelling clinical data, others remain at the preclinical stage with clinical validation still pending.
Targeted treatment strategies supported by clinical dataNTRK targeting in sarcomasThe study of NTRK fusions in sarcomas has become increasingly significant due to the therapeutic implications of targeting these genetic abnormalities with TRK inhibitors. NTRK fusions result from translocations involving one of the three neurotrophic receptor tyrosine kinase genes (NTRK1, NTRK2, NTRK3), leading to the expression of chimeric TRK proteins that have constitutive kinase activity. This aberrant activity promotes oncogenesis through various downstream signaling pathways, making NTRK fusions attractive targets for cancer therapy.
Recent clinical trials and studies have highlighted the efficacy of TRK inhibitors in tumors harboring NTRK fusions, showing significant clinical responses across a diverse set of tumor types, including sarcomas. For instance, the efficacy of larotrectinib and entrectinib, both TRK inhibitors, has been demonstrated in various cancers with NTRK fusions, leading to their approval for use in this context by regulatory bodies like the FDA [51, 52].
Larotrectinib has shown robust efficacy in sarcomas with NTRK gene fusions, with a reported overall response rate (ORR) of 58% in adult patients, and particularly high efficacy in infantile fibrosarcoma [14]. The median progression-free survival (PFS) reached 28.3 months, underscoring the durability of responses, and a 36-month overall survival (OS) rate was impressively high at 77%. This positions larotrectinib as a potent treatment option, especially for pediatric sarcoma patients where it has shown to alter disease course significantly.
Entrectinib provides a broader therapeutic benefit, given its ability to cross the blood-brain barrier, which is advantageous for patients with central nervous system (CNS) involvement. The updated efficacy data from an integrated analysis reveals an ORR of 57.7% in the NTRK fusion-positive sarcoma cohort, with a median duration of response (DoR) of 15.0 months and median OS of 18.7 months [15]. These results highlight entrectinib’s ability to manage diverse sarcoma histologies effectively.
Both drugs have well-documented safety profiles with manageable side effects, making them suitable for long-term management of sarcoma patients harboring NTRK fusions. The choice between larotrectinib and entrectinib may be influenced by specific patient needs, including CNS involvement and previous treatments.
The compelling data from both therapeutic agents emphasize the necessity of routine genomic screening for NTRK fusions in sarcomas to tailor personalized treatment approaches that can significantly improve patient outcomes. This shift towards precision medicine in oncology not only enhances the efficacy of treatment regimens but also minimizes unnecessary exposure to less effective therapies.
EZH-2 inhibitors in epithelioid sarcomaSMARCB1 (SWI/SNF-related matrix‐associated actin‐dependent regulator of chromatin subfamily B member 1), also known as INI1 (integrase interactor 1), serves as a core subunit within the SWI/SNF ATP‐dependent chromatin remodeling complex [53]. Functioning as a potent tumor suppressor gene, INI1 regulates diverse cellular processes, including differentiation and proliferation [54]. Genetic aberrations in INI1, identified in a subset of STS, present a potential therapeutic target.
Loss of INI1 function leads to the upregulation of EZH2, a crucial epigenetic regulator. This dysregulation results in the trimethylation of H3K27 on target genes, leading to their repression. Consequently, this cascade contributes to the activation of various oncogenic signaling pathways, including Sonic Hedgehog, Wnt/β-Catenin, and MYC [54].
The frequency of INI1 loss is notably high (50 to 80%) in epithelioid sarcoma (ES) and other sarcomas displaying epithelioid features, such as malignant peripheral nerve sheath tumors (MPNST) [55, 56].
Tazemetostat (EPZ-6438), a potent and highly selective EZH2 inhibitor, demonstrated promising results in a phase 1 trial evaluating its efficacy and tolerability in advanced solid tumors [57]. Notably, a patient with an INI1-negative malignant rhabdoid tumor exhibited a complete response lasting over 4 years. Subsequent inclusion of patients with similar genetic aberrations showed objective responses or prolonged stable disease, particularly in those with INI1- or SMARCA4-negative solid tumors. In INI1-negative tumors, a basket phase 2 study [16], notably in the epithelioid sarcoma cohort, revealed an ORR of 15% [95% CI 6.9–25.8], with a median follow-up of 59.9 weeks. The median duration of response was not reached, and the overall DCR was 26% [95% CI 15.5–38.5]. The mPFS was 23.7 weeks [95% CI 14.7–25.7], and the mOS was 82.4 weeks [95% CI 47.4-NA].
Tazemetostat stands as a paradigm of effective targeted therapy within a specific sarcoma sub-histotype, leading to an exceptional response and the accelerated approval of an epigenetic drug in January 2020 in the USA, specifically for the treatment of adults and adolescents with locally advanced or metastatic ES not eligible for complete resection.
Furthermore, this drug exhibits potential for combination treatment with ICIs. Noteworthy induction of CD8 T cells in an epithelioid sarcoma patient treated with tazemetostat has been reported [57]. Preclinical models have highlighted the role of EZH2 in immunomodulation [58], and synergies between EZH2 inhibition and ICI efficacy have been demonstrated in various types of solid tumors [59, 60]. These findings lay the groundwork for clinical trials, such as the ongoing phase 2 trial CAIRE [NCT04705818], evaluating the combination of tazemetostat and durvalumab in different solid tumors, including STS and TLS-positive tumors.
Preclinical data have also suggested synergistic activity of EZH2 inhibition in combination with chemotherapy [61]. This combination has been demonstrated to be clinically safe, and a Phase 3 study is currently underway. This study aims to compare tazemetostat + doxorubicin against the current frontline standard treatment, single-agent doxorubicin + placebo, as a first-line treatment in locally advanced unresectable or metastatic ES [17].
MDM2 targeting in well-differentiated and dedifferentiated liposarcomaIn the intricate landscape of STS, the aberrant MDM2 (Mouse Double Minute 2) signaling pathway has become a focal point, particularly in LPS, constituting approximately 20% of STS cases. The well-differentiated (WDLPS) and dedifferentiated (DDLPS) subtypes of LPS, characterized by MDM2 amplification and wild-type TP53 gene status, present a compelling therapeutic target [62].
MDM2, acting as a negative regulator of the tumor suppressor protein p53, exerts its influence through a complex interplay involving the binding of MDM2 to the transcription activation domain of p53, ultimately leading to proteasomal degradation. Strategies to disrupt the MDM2-TP53 interaction have been pursued to restore p53 function, promoting apoptosis, cell cycle arrest, and DNA repair in MDM2-amplified, TP53 wild-type tumors [63].
Traditionally, MDM2-TP53 interaction antagonists, such as nutlins and spiro-oxindoles, have been explored [64, 65]. However, a new class of compounds, MDM2 degraders, has garnered attention for its distinct approach. MDM2 degraders, utilizing the PROTAC (PROteolysis TAgeting Chimeras) technology, go beyond mere inhibition of the MDM2-TP53 interaction [66]. Instead, they induce the degradation of the MDM2 protein itself, leading to a reduction in MDM2 levels within the cell. This dual mechanism of action not only decreases the inhibitory effect of MDM2 on p53 but also enhances p53 activity by reducing MDM2 levels.
Recent trials, such as the MANTRA phase 3 trial [19] assessing milademetan and the ongoing phase 2/3 trial Brightline-01 exploring BI907828 [18], underscore the complexity of therapeutic interventions in LPS. While the MANTRA trial did not meet its primary endpoint, ongoing investigations into BI907828 in DDLPS highlight the evolving landscape of targeted therapies.
Moreover, the pursuit of MDM2 inhibition extends beyond its classical role in apoptosis. Emerging evidence suggests that MDM2’s oncogenic activities encompass broader transcriptional regulation programs, influencing amino acid metabolism, redox homeostasis, and the expression of stress response genes [67, 68]. The exploration of these additional facets holds promise for uncovering novel therapeutic opportunities and expanding our understanding of the intricate molecular landscape associated with MDM2 inhibition in sarcomas.
As the field progresses, the potential for MDM2 inhibitors, whether disrupting the MDM2-TP53 interaction or inducing MDM2 degradation, offers hope for improved outcomes in STS. The nuanced interplay between these inhibitors and the intricate molecular pathways within tumors remains an area of active research, with the goal of refining therapeutic strategies for a more targeted and effective approach in the diverse landscape of soft tissue sarcomas.
Inhibition of gamma-secretase as a therapeutic interventionIn the intricate landscape of tumor biology, the role of gamma-secretase emerges as a critical determinant, influencing cellular processes through the cleavage of various membrane proteins. Among its notable substrates there is the Notch receptor, a key player in cell differentiation, proliferation, and survival [69]. Dysregulation of the Notch signaling pathway, orchestrated by gamma-secretase, has been implicated in the initiation, progression, and metastasis of tumors.
Within this broader context, desmoid tumors, characterized by their local aggressiveness and unpredictable course, present unique challenges in terms of therapeutic interventions [70]. Traditionally, surgical and local treatments have been considered, yet the infiltrative growth of desmoid tumors in specific anatomical locations often necessitates alternative systemic approaches [70, 71].
Nirogacestat, a first-in-class gamma-secretase inhibitor, has recently emerged as a promising addition to the therapeutic arsenal for desmoid tumors. In the DeFi trial, a phase 3 study, [72] nirogacestat demonstrated a substantial improvement in PFS compared to placebo, signifying a paradigm shift in the management of this locally aggressive disease. The observed increase in the ORR in the treatment arm further underscores the clinical significance of gamma-secretase inhibition, with an ORR of 41% in the treatment arm versus 8% in the placebo arm (p < 0.001).
However, the safety profile of nirogacestat does include some concerns that are particularly relevant to this demographic. The drug has been associated with other side effects such as diarrhea, fatigue, nausea, and rash. More serious risks include ovarian dysfunction, which can lead to menstrual irregularities and potential temporary infertility, and liver problems, which require regular monitoring. The decision to use nirogacestat involves weighing these risks against the benefits of effective tumor control. This balance is particularly important for young, active patients who may be concerned not only with the immediate health impact but also with long-term consequences such as fertility and the possibility of chronic conditions.
In conclusion, the role of gamma-secretase in tumor biology takes center stage, and its targeted inhibition, exemplified by nirogacestat, not only addresses the unique challenges posed by desmoid tumors but also contributes to our broader understanding of the intricate interplay between molecular pathways and tumorigenesis. This therapeutic advancement signifies a promising step forward, offering renewed hope for patients with the complexities of locally aggressive tumors.
Targeted treatment strategies supported by preclinical dataCDK9Cyclin-dependent kinase 9 (CDK9) stands at the intersection of critical cellular processes, exerting a profound influence on transcriptional elongation. As a serine/threonine protein kinase, CDK9 orchestrates the phosphorylation and activation of RNA polymerase II subunit RPB1. This cascade of events culminates in the upregulation of pivotal oncogenic genes, such as myeloid cell leukemia-1 (MCL-1) and c-Myc [73]. In the intricate dance of cellular machinery, CDK9 emerges as a central conductor, governing the orchestration of gene expression crucial for cell cycle progression, anti-apoptotic mechanisms, and cellular proliferation.
Within the expansive realm of cancer biology, CDK9 has assumed a role of increasing prominence due to its multifaceted contributions to tumorigenesis. Its involvement in transcriptional regulation positions it as a key player in the uncontrolled cellular growth characteristic of cancer. Beyond merely facilitating transcription, CDK9 has been implicated in the dysregulation of vital pathways, offering a unique therapeutic opportunity [74, 75].
Sarcomas present a compelling battleground for CDK9-targeted interventions [
留言 (0)