
記住我









I hypothesize that our body is able to control the growth of some cancers in a pragmatic and bi-directional switch-like manner. Metabolic syndrome is the most immediate threat to the body’s short-term survival, and our body might induce, promote, or tolerate cancers as a mechanism to consume large amounts of blood glucose through the cancer’s Warburg effect in order to combat this metabolic threat. Conversely, this switch can be reversed to attempt to halt or reverse tumor growth after the metabolic pathology has been resolved. When utilizing this switch greatly depends on the nutritional status of a person: a constant high calorie and carbohydrate diet leads to chronic inflammation with the switch for cancer growth frozen in the on-state to consume as much glucose as possible to counteract the metabolic dysfunction. However, when metabolic dysfunction and chronic inflammation is resolved, for example through a modified diet, the switch for cancer growth is turned off and the body attempts to shut down cancer growth. Our body makes pragmatic and intelligent decisions that address immediate threats first and deals with potential long-term risks after the immediate threat has been eliminated. This review will ask several key questions: Why does a high-glucose diet suppress the immune system? Why does cancer almost always choses a highly inefficient energy metabolism? Why is cancer not contagious? Why does the body allow large blood supplies to the tumor? Why do large tumors suppress smaller tumors? Why to cancers take toxic glucose and turn it into non-toxic lactate without using toxic oxygen? Why cancers are so sensitive to a drop in glucose and normal cells are not? Why is there linear relationship between major cancers and calorie intake?
IntroductionLet food be thy medicine and medicine be thy food. (Hippocrates)
When diet is wrong, medicine is of no use. When diet is correct, medicine is of no need. ∼Ayurvedic proverb.
Cancer is a major health concern in modern human society, with an estimated 600,000 deaths from cancer in 2020 [1]. Cancer progression utilizes several mechanisms, including oncogenic mutations to allow cancer cells to proliferate, expression of the CD-47 protein to defeat the immune system [2], initiate angiogenesis to ensure blood supply, and to enable cell mobility cells to escape the primary tumor site and metastasize to distant organs.
Cancer cells also make a curious metabolic choice: cancer frequently does not utilize oxidative phosphorylation of glucose for energy production, but instead show dramatically increased uptake of glucose, and glycolysis of glucose into lactate, even in the presence of abundant oxygen (Warburg effect [3]).
But perhaps there is also another story: that, under certain conditions, cancer has a purpose. Our body, in the face of an immediate existential threat like metabolic syndrome, has an explicit interest to promote cancer cells to survive in order to pragmatically use the specialized cancer metabolism to combat excessive and toxic glucose levels when the body becomes insulin resistant. The interactions of an organism and cancer cells are complex - but several characteristics of these interactions might suggest that cancer growth is promoted in situations of metabolic syndrome, for example by suppression of the immune system, allowing a large blood supply to the tumor, and impairment of the growth of secondary tumors. Furthermore, the body can reverse this process of promoting cancer growth as soon as the metabolic emergency has been resolved, as cancer cannot survive in a low glucose environment unlike normal cells. I propose that our body uses levels of chronic inflammation as a switch to pragmatically turn cancer growth on and off depending on the short-term survival needs for the organism.
Metabolic syndrome is one of the biggest medical problems in modern society [4]. Progression towards full blown metabolic syndrome is a decade-long process during which the body tries to keep metabolic dysfunction under control. However, with increasing insulin resistance blood sugar levels cannot be controlled anymore, and our body has reached a stage where this decade-long metabolic battle appears to have been lost [4]. In “A new link between diabetes and Cancer” [5] the authors state: “Certain experimental cancers behave more aggressively when animals overeat, and less aggressively when animals are calorically restricted” and “Animals fed a calorie-restricted diet show a strong reduction in plasma glucose levels and prolonged survival. The direct effects of high glucose on tumor cells include increased proliferation and the wiring of the cancer associated signaling pathways.” Another literature example is Wilhelm Brunigns’ contribution to the metabolic treatment of cancer utilizing hypoglycemia [6]: “Ketogenic diets (KD) that are capable of slowing down tumor growth in many animal models. Blood glucose curves between the cancer patients and normal subjects found elevated fasting glucose concentrations for the former, but also abnormal postprandial elevation after a very low carbohydrate meal. KD’s by themselves have been shown to impair glycolytic tumor metabolism in humans … in which tumor lactate levels dropped significantly after only a few days on the diet.”
This communication wants to propose a novel concept in which our body uses its sophisticated set of subsystems and sensors to intelligently and pragmatically anticipate and combat the most direct and dire threat first, while temporarily accepting a state that in any other circumstances would be considered detrimental. Our body is able to control cancer growth in a pragmatic and bi-directional switch-like manner to firstly induce and/or promote cancers as a mechanism to consume large amounts of blood glucose through the cancer’s Warburg effect in order to combat the most immediate threat to the body’s short-term survival, and then reverse this switch to halt or reverse tumor growth after the metabolic pathology has been resolved.
Cancer metabolism is adapted to hypoxic conditions by utilizing glycolysis, and not oxidative phosphorylation, to produce energy [3]. Since glycolysis is a very inefficient energetic mechanism, large amounts of glucose are used by cancer cells to provide the energy needed [3] and thus represents a mechanism by which glucose is removed from circulation. This mechanism has the potential to contribute to counteracting the problem of an ever-increasing insulin resistance and systemic accumulation of glucose. Once glucose levels and metabolic syndrome are normalized with the help of the cancer’s glucose consumption, chronic inflammation is reduced, which in turn will reduce oncogenic signaling from the microenvironment to tumor cells and will slow down or prevent further tumor growth. In this model, our body makes a decision to fight the most immediate threat first by supporting cancer growth through chronic inflammation and associated signaling molecules to ensure survival. Interestingly, new research suggests that dietary restrictions, including fasting and low carbohydrate diets, indeed appear to slow down tumor growth and reduce the size of tumors, suggesting that these life-style choices can be utilized to reduce chronic inflammation and the associated oncogenic signaling from microenvironment to cancer [7]. Since in this scenario survival is not threatened by metabolic syndrome and high blood glucose levels anymore, our body has no further need to support a tumor and thus starves it of the oncogenic signals it requires for growth. Our body appears to have the ability to both induce and restrict cancer growth, depending on the needs to best ensure short-term survival. The potent mechanism utilized for this regulation of cancer growth include inflammation and associated signaling pathways [8]. This novel model presented here proposes that our body makes intelligent and pragmatic choices that, for any given circumstance, represent the best chance for short-term survival, even at the cost of promoting potentially changes that, if not reversed, might be detrimental in the long-term.
Observations Supporting the Proposed ModelCancer Provides a Short-Term Survival Benefit by Metabolizing Large Amounts of GlucoseMost cells in our body utilize oxidative phosphorylation of glucose in mitochondria to produce energy. However, cancer cells feature an altered metabolism with dramatically increased uptake of glucose and glycolysis of glucose into lactate, even in the presence of abundant oxygen (Warburg effect) [3]. Energy production through glycolysis is not efficient and requires far greater amounts of glucose to produce equivalent amounts of the energy produced by oxidative phosphorylation. Indeed, it has been estimated that tumor cells can consume 10–100 times more glucose than normal cells [3]. It is not clear why cancer cells would prefer this inefficient metabolic pathway, even when oxygen for the much more efficient oxidative phosphorylation pathway is abundant. This curious preference of cancer cells could however be advantageous when the most imminent threat to the body’s survival is severe insulin resistance, with an ever-increasing accumulation of glucose threatening the very functioning of vital organs. In this instance, any means to reduce systemic glucose levels would be critical to ensure survival. Promoting cancer growth in such a dire emergency would be a pragmatic and intelligent choice for our body to reduce toxic systemic glucose levels and ensure short term survival.
Glucose Concentrations Correlate With Clinical Cancer OutcomesSystemic glucose levels are closely correlated with cancer growth. High glucose levels have been shown to accelerate cancer cell proliferation in vitro, while glucose deprivation has led to cancer cell apoptosis [7, 9]. All tumors utilize glucose, and the vast majority of the tumors proliferate optimally on excess glucose [10]. Patients with type I and type II diabetes have higher cancer rates [4, 11], and exposure of cancer cells to hyperglycemic conditions leads to the activation of oncogenic pathways [8]. Clinical evidence has shown that lower blood glucose levels in late-stage cancer patients is correlated with better outcomes [7]. Dietary restrictions, including fasting and low carbohydrate diets, have been suggested to slow down tumor growth and reduce the size of tumors [7, 9, 12]. Thus, blood glucose levels and tumor growth appear to be closely correlated. This theory might also explain why exercise reduces the risk of cancer. Regular exercise promotes insulin sensitivity and normal blood glucose levels [13], thus making cancer as a mechanism to reduce toxic levels of glucose unnecessary. Proof-of-principle that hypoglycemia itself can induce tumor regression was provided in 1962 by Koroljow who reported the achievement of a 1-year complete remission in two metastasized cancer patients who were put into an insulin coma (lowest blood glucose reading 22 mg/dl) [14]. In 1941 and 1942, Brunings published two reports and findings that carbohydrate metabolism is a general factor necessary for cancer development [6].
In contrast to normal cells, cancer cells cannot tolerate a low-calorie environment. In a mouse model of human metastatic cancer, after 34 days of five 48-h fasting cycles, tumor size was less than half of that in normally fed mice [9]. Indeed, fasting alone was effective in retardation of the growth of many cancer types, and in some cases was similar to the effect of chemotherapy drugs [9]. Moreover, while fasting was as effective as chemotherapy alone, adding fasting to chemotherapy showed synergistic effectiveness. These studies concluded that the use of fasting is a potential tool to increase the effectiveness of chemotherapy while lowering side effects [9]. Similar results have been obtained for a number of different types of cancers [12], suggesting that normal cells have a defense against the environmental stress of low-calorie intake, but cancer cells do not. Fasting also affects hormones such as thyroid hormone, testosterone, insulin, cholesterol, and C-reactive protein [15, 16, 17], and many cancers are linked to high levels of these proteins and hormones [16, 17]. These observations support the hypothesis presented here that as soon as glucose levels and calorie intake are not problematic anymore, inflammation is reduced, and cancer growth can be slowed or halted, as this function of the tumor is no longer needed. In addition to hyperglycemia, hypercholesterolemia has also been shown to play critical roles in cancer [18]. While hyperglycemia is often driven by lifestyle, i.e., the uncontrolled uptake of sugars, 80% of cholesterol is produced by our body, and only 20% are taken up through our diet. Moreover, autoregulatory mechanisms appear to balance endogenous production when dietary cholesterol intake is increased [18]. Thus, it appears that hyperglycemia is more of a modifiable risk factor when compared to hypercholesterolemia. Nevertheless, metabolic changes other than hyperglycemia, including hypercholesterolemia, undoubtably play important roles in cancer progression as well, and have been shown to be targetable in cancer therapies [18].
Cancers can Evade the Human Immune SystemCancer cells utilize sophisticated masking mechanisms to evade the immune system. Expression of CD47 protein has been found to be common in many cancers [19]. CD47 is normally used as a date code by the body’s own cells in order to prevent young cells from being attacked by the immune system. Aging cells lose CD47 expression and are targeted for elimination by the immune system [19]. Cancer cells utilize this system and express cell surface CD47 to evade attacks by immune cells. Weissman calls CD47 molecule the “Don’t Eat Me” molecule which blinds the immune system to the cancer cell [19]. The model proposed here argues that natural selection would have eliminated this problem unless there are circumstances under which it is beneficial for the body to tolerate and even support growth of a tumor. Late-stage metabolic syndrome would be an example of such a circumstance under which a tumor would be tolerated to contribute to the fight against the immediate metabolic threat.
Tumors can Promote Angiogenesis to Ensure Sufficient Blood SupplyCancers can promote angiogenesis to initiate and maintain vascularization and therefore ensure sufficient blood supply to tumor tissue [20]. This observation is consistent with a functional importance of a tumor to participate in the removal of glucose from circulation - indeed, in order to significantly change systemic glucose levels, it is critical that a good vascularization of the tumor is maintained. The model presented here argues that the body tolerating a large tumor that requires significant resources and blood supply might indicate that there are circumstances when this tumor represents a temporary benefit to the organism.
The Reverse Warburg Effect describes the ability of epithelial cancer cells to manipulate surrounding normal stroma to undergo myofibroblastic differentiation and become tumor-associated stroma, thus creating a cancer-supportive tumor microenvironment (TME) that facilitates further tumor growth and tumor angiogenesis [21]. It has been postulated that this is achieved through secretion of hydrogen peroxide by cancer cells, leading to oxidative stress in surrounding stromal cells and causing their metabolic change to aerobic glycolysis and production and release of high energy metabolites, which subsequently can be utilized by cancer cells to accelerate tumor growth [22]. This Reverse Warburg Effect has been described as inflammation of the tumor microenvironment, and thus would accelerate the chronic inflammation already present in obese patients. The Reverse Warburg Effect would thus decrease excessive blood glucose levels even further by utilizing metabolic changes to aerobic glycolysis in both cancer cells as well as the surrounding TME [23, 22]. Mechanistically, it has been proposed that Caveolin-1 (Cav-1) is a key regulator of the Reverse Warburg Effect and TME phenotype, suggesting that pharmacological targeting of Cav-1 might be a promising avenue for targeted anti-cancer therapies [21].
Tumors Secrete Inhibitors that Suppress Secondary Tumors and MetastasesOne of the most mysterious aspects of cancer biology is the ability of primary tumors to inhibit growth of secondary tumors and metastases [24]. Surgical removal of the primary tumor may stimulate growth of its metastatic secondary tumors [24]. It is not clear why this would be an advantage for the cancer. However, looking at this from the perspective of an organism trying to get the immediate threat of toxic glucose levels under control, this observation could make sense. The primary tumor is supported by the body to help reduce glucose levels, but the organism also wants to make sure that its survival is not compromised by out-of-control metastases formation. This scenario might suggest that our body chooses an intelligent compromise between temporarily tolerating one, but not many tumors, in order to fight metabolic dysfunction, even though multiple tumors would consume more glucose. A single tumor mass that assists in the re-establishment of normal glucose levels is easier to control when the immediate threat of metabolic syndrome has been resolved. In this case, reduced chronic inflammation would reduce pro-tumorigenic signaling from the microenvironment to the tumor and halt or even revert tumor growth, without ill effect on the organism’s survival.
Early Tumor Cell Dissemination and Metastasis FormationLiterature has provided “striking evidence that tumor cells start to disseminate during the initial steps of tumor development that late appearing metastases arise from these early disseminated tumor cells” [25]. However, in the above paragraph it was argued that the primary tumor can inhibit growth of metastasis. This discrepancy can be reconciled by a scenario in which the body wants to support a primary tumor for its metabolic activity and its ability to remove glucose from circulation, but does not want to create a situation in which a significant metastasis burden threatens survival of the organism. Early tumor dissemination and formation of microscopic metastases, the further growth of which is subsequently controlled by the primary tumor, allows a tight control of cancer cell activity to reduce glucose levels without the life-threatening consequences of a large metastasis burden. Importantly, these micro-metastases could function as a backup system in case the primary tumor cannot fulfil its presumed function anymore, for example after surgical removal of the tumor. In this case, the back-up system is activated to allow the micro-metastases to grow and continue to reduce systemic glucose levels.
High Amounts of Glucose Inhibit the Immune SystemIf our body wants to promote a cancer to grow and assist in the reduction of toxic glucose levels, then it would be beneficial if the immune system would be suppressed in situations of metabolic syndrome and excess consumption of sugars and fat. Indeed, the immune system is suppressed when the body ingests larger quantities of both. In the article “Fast Food Fever” the authors state: “In vitro evidence suggest processed, simple sugars also reduce white blood cell phagocytosis and possibly increase inflammatory cytokine markers in the blood.” [26]. Of note, the authors attribute their findings to “glycemic load” of meals rather than sugars themselves. This statement describes a scenario in which sugars decrease immune function, which allows a tumor to grow or survive and, in the model proposed here, would allow the tumor to participate in the reduction of the chronic glycemic load.
The Control of Cancer Cells by Microenvironmental Cues and Inflammation StatusWhile it is widely accepted that DNA damage and mutation in specific genes can drive tumor progression, it also has been shown that microenvironmental cues can promote or inhibit tumor growth. For example, studies showed that transplantation of cancer cells into an embryonic tissue environment cause cancer cells to adopt non-cancerous phenotypes, and normal control of proliferation was re-established [27, 28]. Thus microenvironmental signals can override the phenotypic effects of oncogenic mutations and normalize cell behavior [29]. Vice versa, chronic inflammation and tumor stromal cells have been shown to secrete pro-tumorigenic signaling molecules that can drive tumor progression [8, 30]. These well-documented observations show that our body can utilize signaling mechanism from the cellular microenvironment to control tumor growth bi-directionally, i.e., cancer growth can be induced and accelerated, but also inhibited. This data might also suggest that there could be a benefit for an organism to be able to control cancer growth in this bi-directional manner. This manuscript argues that metabolic syndrome with high systemic glucose levels is such an instance—tumor growth is induced and accelerated through microenvironmental inflammatory signals in order to activate an additional mechanism that can contribute to the reduction in glucose levels. Once glucose is brought under control, chronic inflammation is reduced, which leads to a reduction of pro-tumorigenic signaling and a stop of tumor growth. This proposed mechanism would utilize inflammation and associated signaling events as the principal switch to allow the bi-directional control of cancer cells, even in the presence of oncogenic mutations, to enable an organism to better resolve chronic physiological and metabolic challenges.
Type II Diabetes/Metformin in CancerEpidemiological evidence suggests that type II diabetic patients have a higher risk for cancers of the liver, pancreas, endometrium, colon, rectum, breast and bladder. However, frequency of other cancers appears not to be affected by type II diabetes (TIID) [31], supporting the notion that cancer is a group of highly heterogeneous diseases, and an identical response of all cancers to TIID might therefore not be expected.
Metformin is one of the first pharmacological interventions utilized for TIID patients to reduce blood glucose levels. Consequently, there has been great interest to investigate whether Metformin could be used in the treatment of cancer. Overall, the usefulness of Metformin in cancer treatment has been controversial; evidence from two clinical trials showed no significant improvements [32], while a recent phase II clinical trial of Metformin as a cancer stem cell-targeting agent in ovarian cancer suggested that Metformin-treated tumors showed a decrease in ovarian cancer stem cells and increased sensitivity to cisplatin treatment, as well as improved survival [33]. Laboratory and observational studies have demonstrated a beneficial effect of Metformin in cancer prevention and treatment. For example, metformin improved prognosis and survival rates of diabetic patients with breast, liver, ovarian/endometrial, colorectal and pancreatic cancers [32]. Metformin has also been evaluated in non-diabetic cancer trials. In breast cancer patients, Metformin leads to a significant reduction in Ki67, BRCA1, and cell cycle genes in cancer cells, and also caused a significant increase of breast cancer cells undergoing apoptosis. Further studies seem to indicate that this effect of Metformin is greater in insulin-resistant patients [32], again supporting the hypothesis of this review that functional mechanisms to reduce blood glucose are critical to interfere with cancer growth. Thus, the current limited evidence seems to support the value for further evaluation of Metformin in phase III clinical studies. These studies might well identify specific patient subpopulations with certain cancers and/or oncogenic mutations that might benefit from Metformin treatment. Mechanistically, it is plausible that metformin interferes with cancer growth due to its upregulation of AMP kinase (AMPK), which leads to a block of mTOR and impairment of angiogenesis as well as cell growth and proliferation, key components of cancer progression [32].
Cancer CachexiaCancer cachexia is a wasting syndrome characterize by weight loss anorexia, asthenia and anemia. Interestingly, tumor growth is not switched off in cancer cachexia, despite the extremely low nutritional state [34]. It is possible that in pathological end stage conditions, including cancer cachexia, the bi-directional switch proposed in this manuscript cannot be activated because the associated severe chronic systemic inflammation cannot be resolved anymore by endogenous mechanisms, thus leaving the proposed cancer switch in the “on” position. Interestingly, newer studies promote anti-inflammatory agents and low concentration Insulin treatments for cancer cachexia [34], suggesting that in extreme conditions pharmacological help is required to resolve inflammation and help to push the switch back into the “off” position.
Cancers in Conditions of Severe Nutritional DeficitsSeveral studies have demonstrated a protective effect of moderate calorie restriction on cancer incidence [35]. In contrast, extreme and severe calorie and nutrient restrictions, as experienced during famines and during the Holocaust, have been shown to significantly increase cancer rates [35]. On first sight, this might disagree with the hypothesis of this review that our bodies promote cancer if glycemic load is out of control, and then reduce cancer growth once blood glucose levels have been normalized. However, the Holocaust was associated with critical and life-threatening restrictions in nutrients, extreme psychological stress and violence, and large-scale exposure to the elements and infections [35]. All of these factors are known to have significant effects of metabolic dysregulation, immune function, and subsequently increased cancer rates. Moreover, there might be a long-term effect as well: the drastic slowing of metabolic rates during a famine makes the body more vulnerable to excessive weight gain and obesity once the famine ends, which then would increase the risk of hyperglycemia and cancers. Thus, the increase of cancer rates in survivors of famines and the Holocaust is most likely multifactorial and might not necessarily disagree with the hypothesis put forward in this review.
SummaryThis concept paper proposes a novel hypothesis that envisions cancer as a tool that can be controlled intelligently and pragmatically by an organism in a switch-like bi-directional manner depending on physiological or pathological needs. In the context of metabolic syndrome, severe insulin resistance, and toxic systemic glucose levels, our body will attempt to activate every mechanism possible to ensure short-term survival. One of these mechanisms might be the induction of tumor growth, in order to capitalize on the metabolic specificity of cancer cells to utilize large amounts of glucose for their energy need and thus assist in the body’s attempts to remove glucose from circulation. When the immediate metabolic emergency has been resolved and the tumor is no longer needed, the body can again engage this bi-directional switch to try to halt or reverse tumor growth to avoid long-term detrimental effects (Figure 1). I propose that this unique bi-directional switch is the pragmatic initiation and reduction of inflammation and associated signaling pathways. Of note, cancer might not be the only bi-directional tool activated to reduce glucose level, and subsequently needs to be reversed and inactivated again. For example, the covalent attachment of glucose to many different proteins to form Advanced Glycation End products (AGEs) might also be an emergency mechanism to increase cellular uptake of glucose to remove glucose from circulation and thus ensure short-term survival during metabolic syndrome. Like cancer, AGEs are detrimental to an organism’s long-term survival, have been shown to accumulate in conditions of chronic inflammation, and also promote tumor growth [36]. One could view AGEs as synergistic activators of tumor growth in the context of type II diabetes and metabolic syndrome. AGE molecules will be eliminated through the normal protein elimination pathways, including proteasome and autophagy pathways. Provided that the metabolic emergency has been resolved and systemic glucose levels have returned to normal, new proteins will not be glycated and thus can fulfil their normal function without the negative long-term effects of AGEs. I believe that the evidence presented here supports my hypothesis that our body might be able to pragmatically use and control temporary pathological states like cancer and AGEs to its advantage to resolve short-term threats, including metabolic syndrome, and ensure survival of the organism. It is my hope that this hypothesis might help open novel avenues for intellectual and experimental exploration in order to exploit the pragmatic control of pathological states for better health outcomes.
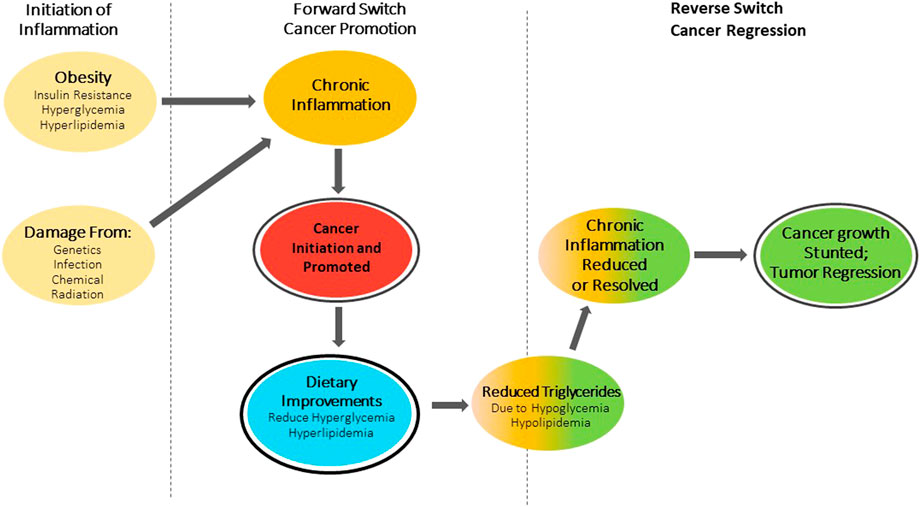
FIGURE 1. Switch theory of cancer. Obesity and associated Insulin resistance causing hyperglycemia and hyperlipidemia or damage from: Genetics, Infection, Chemicals and Radiation initiate a forward switch that leads to chronic inflammation. This inflammation promotes cancer growth. Dietary improvements in metabolic health will trigger a reverse switch, reducing or resolving chronic inflammation and stunting cancer growth and/or lead to tumor regression.
Author’s NoteJC is an independent researcher, and the CEO/ founder of Applied Interconnect Inc. and is a 30-year veteran of aerospace engineering. He views the body as a sophisticated set of subsystems and sensors trying to anticipate its’ threats and fuel needs in a complex environment.
Author ContributionsThe author confirms being the sole contributor of this work and has approved it for publication.
Conflict of InterestJC is the owner of Applied Interconnect Inc. and is employed at the company.
AcknowledgmentsI wish to thank Michael Klüppel, PhD, of MKPhD Scientific Consulting, for editing and proofreading, as well as helpful scientific discussions and advice during the preparation of this manuscript. Without his help the publication of the paper would not have been possible. Further I wish to thank my daughter, Kristina, BS BioEngineering, who contributed to this paper with her knowledge, wisdom, and joyfulness of life and love.
References1. Siegel, RL, Miller, KD, and Jemal, A. Cancer Statistics. CA: A Cancer J Clinicians (2020) 70(1):7–30. doi:10.3322/caac.21590
CrossRef Full Text | Google Scholar
2. Bissell, MJ, and Hines, WC. Why Don't We Get More Cancer? A Proposed Role of the Microenvironment in Restraining Cancer Progression. Nat Med (2011) 17(3):320–9. doi:10.1038/nm.2328
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Harding, JL, Shaw, JE, Peeters, A, Cartensen, B, and Magliano, DJ. Cancer Risk Among People with Type 1 and Type 2 Diabetes: Disentangling True Associations, Detection Bias, and Reverse Causation. Diabetes Care 2015;38:264-270. Cancer Risk Among People Type 1 Type 2 Diabetes: Disentangling True Associations, Detection Bias, Reverse Causation Diabetes Carediabetes Care (2015) 3838(4):264734–2705. doi:10.2337/dc15-er04a
CrossRef Full Text | Google Scholar
5. García-Jiménez, C, Garcia-Martinez, JM, Chocarro-Calvo, A, and De la Vieja, A. A New Link between Diabetes and Cancer: Enhanced WNT/β-catenin Signaling by High Glucose. J Mol Endocrinol (2014) 52(1):R51–66. doi:10.1530/jme-13-0152
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Klement, RJ. Wilhelm Brünings' Forgotten Contribution to the Metabolic Treatment of Cancer Utilizing Hypoglycemia and a Very Low Carbohydrate (Ketogenic) Diet. J Traditional Complement Med (2019) 9(3):192–200. doi:10.1016/j.jtcme.2018.06.002
CrossRef Full Text | Google Scholar
7. Luo, J, Chen, YJ, and Chang, LJ. Fasting Blood Glucose Level and Prognosis in Non-small Cell Lung Cancer (NSCLC) Patients. Lung Cancer (2012) 76(2):242–7. doi:10.1016/j.lungcan.2011.10.019
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Lee, C, Raffaghello, L, Brandhorst, S, Safdie, FM, Bianchi, G, Martin-Montalvo, A, et al. Fasting Cycles Retard Growth of Tumors and Sensitize a Range of Cancer Cell Types to Chemotherapy. Sci Transl Med (2012) 4(124):124ra27. doi:10.1126/scitranslmed.3003293
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Roberts, CK, Hevener, AL, and Barnard, RJ. Metabolic Syndrome and Insulin Resistance: Underlying Causes and Modification by Exercise Training. Compr Physiol (2013) 3(1):1–58. doi:10.1002/cphy.c110062
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Tantamango-Bartley, Y, Jaceldo-Siegl, K, Fan, J, and Fraser, G. Vegetarian Diets and the Incidence of Cancer in a Low-Risk Population. Cancer Epidemiol Biomarkers Prev (2013) 22(2):286–94. doi:10.1158/1055-9965.epi-12-1060
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Akhmedkhanov, A, Zeleniuch-Jacquotte, A, and Toniolo, P. Role of Exogenous and Endogenous Hormones in Endometrial Cancer: Review of the Evidence and Research Perspectives. Ann N.Y Acad Sci (2001) 943:296–315. doi:10.1111/j.1749-6632.2001.tb03811.x
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Lee, CH, Woo, Y, Wang, Y, Yeung, C, Xu, A, and Lam, K. Obesity, Adipokines and Cancer: an Update. Clin Endocrinol (Oxf) (2015) 83(2):147–56. doi:10.1111/cen.12667
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Pavlides, S, Whitaker-Menezes, D, Castello-Cros, R, Flomenberg, N, Witkiewicz, AK, Frank, PG, et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle (2009) 8(23):3984–4001. doi:10.4161/cc.8.23.10238
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Liang, L, Li, W, Li, X, Jin, X, Liao, Q, Li, Y, et al. Reverse Warburg Effect of Cancer-Associated Fibroblasts (Review). Int J Oncol (2022).
23. Pavlides, S, Tsirigos, A, Vera, I, Flomenberg, N, Frank, PG, Casimiro, MC, et al. Loss of Stromal Caveolin-1 Leads to Oxidative Stress, Mimics Hypoxia and Drives Inflammation in the Tumor Microenvironment, Conferring the “Reverse Warburg Effect”: A Transcriptional Informatics Analysis with Validation. Cell Cycle (2010) 9:2201–19. doi:10.4161/cc.9.11.11848
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Janssen, LME, Ramsay, EE, Logsdon, CD, and Overwijk, WW. The Immune System in Cancer Metastasis: Friend or Foe? J ImmunoTherapy Cancer (2017) 5(1):79. doi:10.1186/s40425-017-0283-9
CrossRef Full Text | Google Scholar
27. Kulesa, PM, Kasemeier-Kulesa, JC, Teddy, JM, Margaryan, NV, Seftor, EA, Seftor, REB, et al. Reprogramming Metastatic Melanoma Cells to Assume a Neural Crest Cell-like Phenotype in an Embryonic Microenvironment. Proc Natl Acad Sci U S A (2006) 103(10):3752–7. doi:10.1073/pnas.0506977103
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Kasemeier-Kulesa, JC, Teddy, JM, Postovit, LM, Seftor, EA, Seftor, RE, Hendrix, MJ, et al. Reprogramming Multipotent Tumor Cells with the Embryonic Neural Crest Microenvironment. Dev Dyn (2008) 237(10):2657–66. doi:10.1002/dvdy.21613
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Costa, FF, Seftor, EA, Bischof, JM, Kirschmann, DA, Strizzi, L, Arndt, K, et al. Epigenetically Reprogramming Metastatic Tumor Cells with an Embryonic Microenvironment. Epigenomics (2009) 1(2):387–98. doi:10.2217/epi.09.25
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Iyengar, NM, Gucalp, A, Dannenberg, AJ, and Hudis, CA. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J Clin Oncol (2016) 34(35):4270–6. doi:10.1200/jco.2016.67.4283
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Brown, JR, Chan, DK, Shank, JJ, Griffith, KA, Fan, H, Szulawski, R, et al. Phase II Clinical Trial of Metformin As a Cancer Stem Cell-Targeting Agent in Ovarian Cancer. JCI insight (2020) 5(11):e133247–12. doi:10.1172/jci.insight.133247
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Lundholm, K, Körner, U, Gunnebo, L, Sixt-Ammilon, P, Fouladiun, M, Daneryd, P, et al. Insulin Treatment in Cancer Cachexia: Effects on Survival, Metabolism, and Physical Functioning. Clin Cancer Res (2007) 13(9):p2699–2706. doi:10.1158/1078-0432.ccr-06-2720
CrossRef Full Text | Google Scholar
35.Boker. Increased Cancer Indidence in Holocaust Survivors and Implications for Survivors of Other Extreme Events. Expert Rev Anticancer Ther (2018) 18:10591062. doi:10.1080/14737140.2018.1521274
CrossRef Full Text | Google Scholar
36. Garay-Sevilla, ME, Gomez-Ojeda, A, Gonzalez, I, Luevano-Contreras, C, and Rojas, A. Contribution of RAGE axis Activation to the Association between Metabolic Syndrome and Cancer. Mol Cel Biochem (2021) 476:1555–73. doi:10.1007/s11010-020-04022-z
留言 (0)