Study design and patients
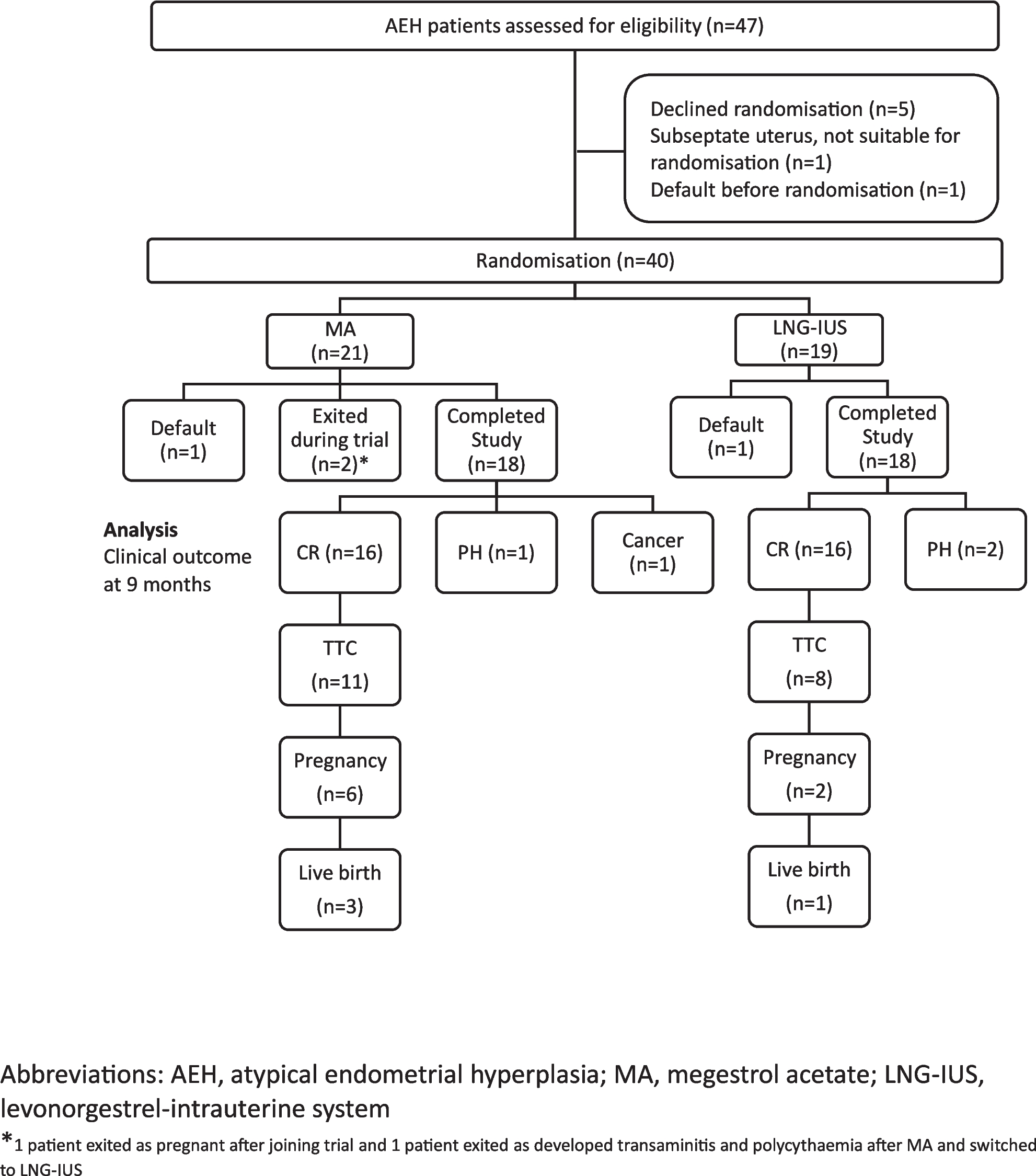
The study was a multi-centre, open-label, randomised controlled phase II trial (NCT05492487) with two study arms comparing MA to LNG-IUS. Patients were recruited from January 3, 2020 to July 14, 2023, and were on follow-up from January 3, 2020 to January 8, 2024. The trial was conducted in the three medical centres in Singapore: KK Women’s and Children’s Hospital, Singapore General Hospital and National University Hospital. This study was supported by the Singhealth AM grant (Grant No.: AM/CT004/2020).
Eligible patients with AEH who were aged of 21–40 years and had strong desires to preserve fertility were included in the study. Patients with previous or current history of endometrial cancer, or who were already on treatment for AEH were excluded. Patients who were unable to be randomised (i.e., contraindications to either MA or LNG-IUS) were excluded.
The study was approved by the Singhealth Centralised Institutional Review Board (CIRB) (CIRB Ref: 2019/2551) and registered with the Health Science Authority (HSA) (License No.: CTA1900087) (https://eservice.hsa.gov.sg/prism/ct_r/enquiry.do?action=loadSpecificDetail) on September 5, 2019, prior to the recruitment of the first patient, as per our national requirement. The trial was registered retrospectively on ClinicalTrials.gov (ID: NCT05492487) on April 7, 2022 (https://clinicaltrials.gov/study/NCT05492487). This trial was only registered retrospectively on ClinicalTrials.gov when the study team found out about the need for registration on an approved registry as a requirement for publication. There were no changes to protocol our endpoints during the entire conduct of the study. All patients were fully informed of the benefits and risks of this clinical trial and provided written informed consent.
Randomisation and masking
Patients were allocated (1:1) to one of two treatment arms: MA vs. LNG-IUS using randomisation envelopes. A total of 60 randomisation envelopes divided over the 3 institutions were created at the start of the trial. The allocation sequence was concealed from the study team enrolling and assessing participants in opaque and sealed envelopes. The study was open labelled, and all patients and study physicians were aware of the treatment assignment.
Procedures
In literature, the dose of megestrol acetate used for the treatment of atypical endometrial hyperplasia and/or endometrial adenocarcinoma was 160–320 mg per day [8, 21]. In the latest ESGO/ESHRE/ESGE guidelines on fertility-sparing treatment of endometrial cancer, the recommended dose was-320 mg per day [22]. As our study group involved atypical endometrial hyperplasia, we used the starting dose of 160 mg per day.
The patients in the MA arm received continuous oral megestrol acetate 160 mg once daily and the patients in the LNG-IUS arm underwent LNG-IUS (containing LNG 52 mg) insertion.
All patients underwent a hysteroscopy D&C for the first diagnosis before joining the trial to exclude the presence of concomitant endometrial cancer. Pathologic diagnosis was confirmed by the pathologists in each institution, according to the World Health Organization (WHO) pathological classification (2014) [23].
Endometrial evaluations were performed every 3 months to evaluate treatment response after initiation of the treatment until complete response (CR), by a member of the study team. Endometrial evaluation was done by hysteroscopy and targeted endometrial biopsies, or by outpatient endometrial biopsy (without hysteroscopy). The LNG-IUS was kept in situ during the biopsy. If dislodged during the biopsy, a new LNG-IUS would be inserted.
Treatment response was categorized as follows: (1) complete regression (CR): histology showing resolution of endometrial hyperplasia; (2) persistent hyperplasia (PH): persistent endometrial hyperplasia (non-atypical or atypical); (3) progression disease (PD): progression of disease to endometrial cancer. The treatment was continued until CR was achieved, or at a maximum duration of treatment of 9 months in the trial. Treatment was discontinued when patients experienced unacceptable side effects or when patients were keen to switch to another medical treatment or surgery (hysterectomy). Patients with PD would be referred to the gynaecologic oncologist for further management. Patients treated beyond 9 months would exit the study. The decision to continue medical treatment or opt for hysterectomy would be discussed in this group of patients.
Patients in the trial who achieved CR and were keen to achieve fertility would be reviewed by the reproductive medicine unit. Fertility options including ovulation induction, intrauterine insemination with or without superovulation, and in vitro fertilisation would be discussed.
For CR patients with no immediate plans for conception, patients were offered cyclical dydrogesterone, oral contraceptive pills, or insertion of LNG-IUS for prevention of disease recurrence.
Ultrasonography, physical review and/or endometrial biopsies would be performed every 6 months to 1 year to assess for recurrence.
Data on age, presenting complaints, presence of polycystic ovarian syndrome (based on the Rotterdam Criteria), presence of diabetes mellitus, impaired glucose tolerance (IGT) or impaired fasting glucose based on the World Health Organisation (WHO) diagnostic criteria, comorbidities and ultrasonography findings were collected during the study. The patients’ height and weight were collected after every 3 months of treatment (3-month, 6-month and 9-month).
All patients were followed up from the date of treatment initiation to January 8, 2024.
Outcomes
The primary endpoints were the CR rate at 9 months and the time taken for CR. Secondary endpoints were the side effects, patient acceptability, fertility outcomes (pregnancy, miscarriage and live birth rates) and recurrence.
Statistical analysis
A previous meta-analysis on retrospective data has shown improved treatment outcome with LNG-IUS compared to oral progestogens [9]: pooled regression of 90% vs. 69% respectively.
The sample size of 60 was calculated to detect a non-inferiority margin difference between the group proportions of − 0.05. The reference group proportion was 0.70. The treatment group proportion is assumed to be 0.65 under the null hypothesis of inferiority. The power was computed for the case when the actual treatment group proportion was 0.90. The test statistic used was the one-sided Z test (un-pooled). The significance level of the test was 0.05. Based on retrospective clinical data of AEH patients in our centres, we expected to recruit 60 patients over 2 years. However, due to the finite duration of research funding and the restrictions and difficulty of recruitment during the COVID-19 pandemic, the final recruitment number was 40 patients.
Continuous variables were represented as median and interquartile range, where applicable, and were compared using the Mann–Whitney U test. Categorical variables were represented as frequency and percentage, and compared using Pearson’s chi-square test or Fisher’s exact test, where applicable. The significance was set as 2-sided P < 0.05. All statistical analyses were performed using Stata 18 (Stata Corporation, USA).
Role of funding source
The funding body had no role in the study design, data collection, data interpretation, data analysis, or drafting or editing of this manuscript.






留言 (0)