
記住我

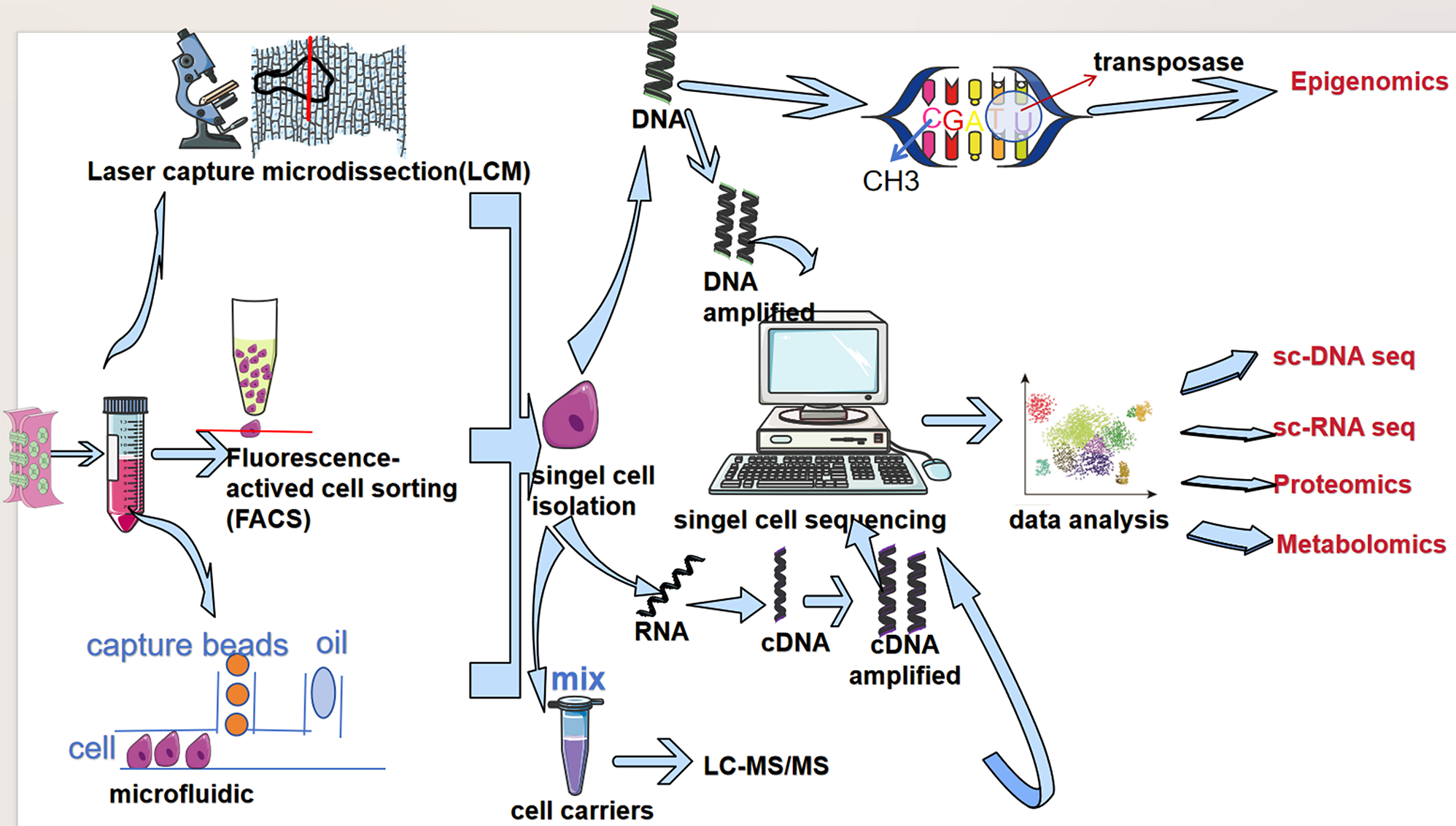



The analysis process of the article was represented in Fig. 1. With stringent quality control metrics in place prior to pre-processing, we managed to obtain 36,680 high-quality cellular samples from 7 BLCA tissues (Supplementary Fig. 1A). The number of genes and the sequencing depth were found to have a strong positive correlation after normalization, with a Pearson correlation coefficient of 0.92 (Supplementary Fig. 1B). With a resolution of 0.8, 20 clusters were identified (Fig. 2A). The cell type annotation was based on the markers that have been reported in literature (Supplementary Fig. 1C). Thus, clusters 15, 17, and 19 were identified as immune cells; cluster 16 was identified as stromal cells; remaining clusters were identified as epithelial cells (Fig. 2B and C), which were consistent with previous classification results [9]. Based on single-cell transcriptome data from BLCA samples, without relying on normal cells as controls, 3 and 8 clusters (a total of 4962 cells) were identified as malignant epithelial cells (ECs) using CopyKAT (Fig. 2D, Supplementary Fig. 1D). Compared to epithelial cells and normal clusters, the 3 and 8 clusters showed high expression of BLCA markers (Supplementary Fig. 1E). The results of functional enrichment analysis for differentially expressed genes between malignant ECs and ECs demonstrated that some signaling pathways associated with carcinogenesis were activated, including VEGFA and TP53 signaling pathways (Supplementary Fig. 1F). These results supported that 3 and 8 clusters were cancerous clusters.
Fig. 1
The flowchart of analysis
Fig. 2
Identification of cell subtypes. (A) 20 clusters are identified at a resolution of 0.8. (B) Markers that identify different cells are expressed. We identify three major cell clusters: epithelial cells, stromal cells, and immune cells. (C) The UAMP plot displays the distribution of epithelial cells, stromal cells, and immune cells. (D) The results of CopyKAT for predicting malignant ECs. 3 and 8 clusters are considered malignant ECs.
Identification of stem cells cluster for BLCAWe drew 3 and 8 clusters of cells from the total cell population. They were subjected to the process of normalization, dimension reduction, and clustering once again. With a resolution of 0.25, 6 clusters were identified (Fig. 3A). The main differential genes of each cluster were shown in the Fig. 3B. GO and KEGG enrichment analysis for the 6 clusters showed that genes in cluster 1 (C1) were enriched in the signaling pathways that were related to chemical carcinogenesis- reactive oxygen species, glutathione metabolism et al.; genes in cluster 2 (C2) were enriched in the signaling pathways involved in pyrimidine metabolism, platinum drug resistance, cell cycle, DNA replication et al.; genes in cluster 3 (C3) were enriched in the signaling pathways implicated in mineral absorption, antigen processing and presentation et al.; genes in cluster 4 (C4) were enriched in the signaling pathways that were associated with leukocyte transendothelial migration, adhere junction, bladder cancer et al.; genes in cluster 5 (C5) were enriched in the signaling pathways that were connected to p53 signaling pathway activation, HIF − 1 signaling pathway activation, cellular senescence et al.; genes in cluster 6 (C6) were enriched in the signaling pathways that were related to antigen processing and presentation, Th1 and Th2 cell differentiation et al. (Fig. 3C, Supplementary Fig. 2A-2 F). The pseudotime trajectory analysis of malignant ECs was performed with Monocle2 to elucidate the developmental trajectory of the ECs (Fig. 3D) and to show that different clusters were at different stages of differentiation (Fig. 3E). Based on this finding, it can be inferred that C2 or C1 were the initial stages of differentiation in ECs. Furthermore, CytoTRACE revealed that C2 was the initiator in the predicted trajectory with higher differentiation potential, and C5 and C3 were the terminal states in the differentiation trajectory (Fig. 3F and G). Thus, C2 was identified as the cancer stem cells for BLCA for further analysis.
Fig. 3
Cell differentiation trajectory. (A) 6 clusters of malignant ECs are identified with a resolution of 0.25. (B) Some of positively differential genes in each cluster. (C) Display the differential genes and KEGG pathway enrichment results of each cluster simultaneously. (D) A color-coded pseudotime plot depicts the differentiation trajectory of malignant ECs in BLCA. (E) Differentiation trajectory of malignant ECs in BLCA for sub-cell types. (F-G) CytoTRACE predicts the cell differentiation potential of malignant ECs in BLCA, and identified C2 as the cancer stem cell cluster
The characterization of the stem cell landscape of BLCAFirstly, we investigated the activity of transcription factors (TFs) and the activation of main pathways, and discovered that in the C2 cluster, E2F1, E2F2, E2F3, E2F4, and TFAM were significantly activated (Fig. 4A, Supplementary Fig. 3A). PI3K and MAPK signaling pathways were involved in regulating the biological functions of the C2 cluster (Fig. 4B, Supplementary Fig. 3B). Secondly, through metabolic analysis, we were able to identify the main metabolic pathways in C2 cluster, including butanoate metabolism, citrate cycle (TCA cycle), fatty acid degradation and elongation et al. (Fig. 4C, Supplementary Fig. 3C), indicating changes in metabolism of CSCs. Thirdly, we drew 16 cluster (stromal cells) of cells from the total cell population. It was subjected to the process of normalization, dimension reduction, and clustering once again. With a resolution of 1, 4 clusters were identified (Supplementary Fig. 3D). According to the expressed markers of cells, clusters 0, 1, and 3 in stromal cells were regarded as cancer-associated fibroblasts (CAFs), while cluster 2 was considered endothelial cells. Based on the expressed markers of CAFs, clusters 0, 1, and 3 were regarded as inflammatory CAFs (iCAFs, markers: CXCL14, MT2A), myofibroblast (myCAFs, markers: RGS5, ACTA2, MYL9), and antigen-presenting CAFs (apCAFs, markers: CD74, HLA-DRA, and HLA-DPA1), respectively (Fig. 4D). The differential genes in each CAFs cluster were showed using heatmap plot (Supplementary Fig. 3E). Then, 15 cluster was considered as myeloid cells (markers: CD14, CD68), while 17 and 19 clusters were regarded as T cells (markers: CD3D, CD3E). The final clusters and annotations can be found in Fig. 4E. The differential gene expression of each cluster cell group was shown in Supplementary Table 1. CellChat analysis showed diverse and distinct interactions among these cell types (Fig. 4F). There were different ligand-receptor modes between C2 and CAFs, as well as between immune cells (Fig. 4G). The secretion of APP, CD99, COL1A1, COL6A1, COL6A3, FN1, MDK, in CAFs, was primarily directed toward CD74, CD99, SDC1, SDC4 receptors on the surface of C2. SPP1 was mainly produced by myeloid cells, which acted on the (ITGAV + ITGB1) receptor on the surface of C2. The MDK secreted by T cells was predominantly directed at the SDC4 and (ITGA6 + ITGB1) receptors on the surface of C2. CellChat analysis of signaling pathways demonstrated that MK, THBS, ANGPTL, VISFATIN, JAM, and ncWNT signaling pathways from tumor microenvironment were involved in regulating the characteristics of C2 (Fig. 4H, Supplementary Fig. 3F). These results demonstrated that modulating the characteristics of CSCs was complex and involved the alternation of multiple molecules and signaling pathways, and the role of CAFs and immune cells in the process should be given more attention.
Fig. 4
Examining the stem cell landscape. (A) The activity of TFs in C2 and other clusters in BLCA. (B) The activation of main pathways in C2 and other clusters in BLCA. (C) The changes in metabolism of C2 and other clusters in BLCA. (D) Stromal cells are subdivided into iCAF, myCAF, apCAF, and endothelial cells (upper). The major markers expressed in iCAF, myCAF, and apCAF (low). (E) The final clusters and annotations for BLCA. (F) CellChat analysis displays communications among these cell types. The impact is greater with a thicker line. (G) CellChat is used to determine selected ligand-receptor interactions between immune cells, CAFs, and C2. The size of each circle is what indicated P-values. The color gradient is a representation of the level of interaction; blue and red colors are the smallest and largest values, respectively. (H) The heatmap exhibits the incoming and outgoing signaling patterns in C2, CAFs, and immune cells. The deeper color indicates more weight and strength of the regulation of the signaling pathway
Integrative construction of a consensus CSCLPI based on machine learningThe differential genes in C2 were abstracted (Supplementary Table 2) and the expression matrices of genes with adjusted P < 0.05 in the TCGA cohort were obtained.
We have performed functional enrichment analysis for the 341 genes to investigate their biological signatures. As shown in the Supplementary Fig. 2B, the results demonstrated that they were mainly involved in the regulation of cell cycle et al. We conducted a univariate Cox analysis and uncovered 29 potential CSCs-related genes that have a significant correlation with the prognosis of BLCA (Supplementary Table 3). The integration of machine learning was used to study the 29 potential CSCs-related prognostic genes. 99 prediction models were fitted in all cohorts by using the leave-one-out cross-validation framework. The results of calculating the C-index of each model across all cohorts were presented in Fig. 5A. Accordingly, the prognostic model developed by the RSF method was found to be the optimal model with the highest average C-index of 0.659 (Fig. 5A). The importance of variables was also evaluated (Fig. 5B), and the variables that contributed the most to the model were selected, including LSM3, S100A10, SPINT1, SPTSSB, and VPS29. A multivariate Cox analysis was conducted on five genes and it was determined that S100A10 (HR:1.90, 95%CI: 1.12 to 3.21, P = 0.016) and SPINT1 (HR: 2.25, 95%CI: 1.36 to 3.70, P = 0.001) were significantly associated with the prognosis of BLCA (Fig. 5C). The CSC-like prognostic index (CSCLPI) was constructed as follows: CSCLPI = (-0.498 * expression of LSM3) + (0.645 * expression of S100A10) + (0.811 * expression of SPINT1) + (-0.269 * expression of SPTSSB) + (-0.472 * expression of VPS29). TCGA samples were classified into the high CSCLPI and low CSCLPI groups using the median value of 1.045 as the cutoff value of the model. The KM survival analysis revealed a difference in survival between the CSCLPI subgroups (Fig. 5D). In the TCGA-BLCA training cohort, the area under the ROC curve based on the CSCLPI for predicting the 1-, 2- and 3-year survival was 0.749, 0.696 and 0.65, respectively (Fig. 5E). In the external validation cohorts, CSCLPI was also developed based on the five genes, and 1.045 was considered as the cutoff value to classify samples. GSE13507 and GSE48075 cohorts had distinct survival differences based on KM survival analysis (Fig. 5F), but GSE32894 and GSE48276 cohorts did not (Fig. 5F). In GSE13507, the area under the ROC curve for predicting the 1-, 2- and 3-year survival was 0.75, 0.627 and 0.656, respectively. In GSE32894, it was 0.765, 0.755 and 0.706, respectively.
Fig. 5
Construction and validation of CSCLPI. (A) The C-index of a total of 99 prediction models from all datasets is based on 10 integrative machine learning algorithms using the LOOCV framework. The RSF method was utilized to construct the model, which had the highest average C-index of 0.659. (B) Ranking of variables’ importance. (C) The results of multivariate Cox regression analysis, and independent prognostic factors were determined. (D) The result of the KM survival analysis of CSCLPI in the TCGA cohort. (E) The results of the ROC curve for CSCLPI in predicting 1-year, 2-year, and 3-year survival in the TCGA cohort. (F) The results of the KM survival analysis of CSCLPI in the validation cohorts. (G) The results of the ROC curve for CSCLPI in predicting 1-year, 2-year, and 3-year survival in the validation cohorts
In GSE48075, it was 0.652, 0.733 and 0.815, respectively. In GSE48276, it was 0.615, 0.552 and 0.513, respectively (Fig. 5). The 1-year AUC value of CSCLPI was compared against those of a stem cell signature [36] and an EMT-related gene signature [37] in BLCA. In Supplementary Fig. 4, it is shown that the 1-year AUC value of a stem cell signature and an EMT-related gene signature were 0.71 and 0.659, respectively, whereas the CSCLPI was 0.749. These findings demonstrated the good performance of the CSCLPI in predicting the prognosis of patients with BLCA.
Differences in mRNAsi, gene mutation, small molecular drug sensitivity, and functional enrichment between CSCLPI subgroupsThe OCLR algorithm was employed to calculate mRNAsi of each BLCA patient by analyzing their gene expression profiles. It was discovered that mRNAsi was significantly higher in the low CSCLPI group (P < 0.001) (Fig. 6A), and mRNAsi was negatively linked with the CSCLPI score (Spearman correlation = − 0.32, P < 0.001) (Fig. 6B). According to KM analysis, patients with high-mRNAsi in BLCA had a longer OS than those with low-mRNAsi (Fig. 6C).
Fig. 6
Certain characteristics analysis for CSCLPI subgroups. (A) The comparison of the mRNAsi between CSCLPI subgroups. ***: p < 0.001. (B) The Spearman correlation of the CSCLPI score with mRNAsi. (C) The KM survival analysis showed that BLCA patients with higher mRNAsi had a better prognosis. (D) The waterfall plot demonstrated mutational genes in the high CSCLPI group (upper) and in the low CSCLPI group (low). (E) Summary for mutation in the high CSCLPI group (upper) and in the low CSCLPI group (low). (F) Small molecular drug sensitivity analysis between CSCLPI subgroups. (G) GO analysis of differential genes between CSCLPI subgroups for the top ten enrichment. (H) KEGG analysis for the top ten enrichment
The results from gene mutation analysis demonstrated that the gene mutation frequency in the high CSCLPI group was higher than that of the low CSCLPI group (95.89% vs. 94.59%). The top 10 mutational genes in the high CSCLPI group contain TTN, TP53, ARID1A, MUC16, KMT2D, KDM6A, RYR2, SYNE1, EP300, and KMT2C (Fig. 6D), which also have a mutation rate of over 20%. The top 10 mutational genes in the low CSCLPI group comprise of TP53, TTN, KMT2D, MUC16, KDM6A, SYNE1, ARID1A, PIK3CA, FLG, and KMT2C (Fig. 6D), which also have a mutation rate of over 20%. The most common mutation type in both groups was missense-mutation, followed by nonsense mutation and splice site (Fig. 6E). The correlation between mutated genes was analyzed in CSCLPI subgroups (Supplementary Fig. 5A). It was found that in the high CSCLPI group, RTK − RAS, NOTCH, Hippo and TP53 signaling pathways were highly and abnormally activated, while in the low CSCLPI group, RTK − RAS, NOTCH, Hippo, TP53, WNT and PI3K signaling pathways were highly and abnormally activated (Supplementary Fig. 5B).
We picked 10 drugs with the most significant P-value based on the results of small molecular drug sensitivity analysis. The findings showed that high CSCLPI group was more sensitive to belinostat, blebbistatin, brivanib, and daporinad, while low CSCLPI group was more sensitive to canertinib, dasatinib, gefitinib, ibrutinib, lapatinib, and myriocin (Fig. 6F).
Functional enrichment was carried out on the differential genes (low-CSCLPI vs. high-CSCLPI) between CSCLPI subgroups. The results of the GO analysis showed that the up-regulated genes were involved in regulation of extracellular matrix (Fig. 6G). KEGG analysis demonstrated that the up-regulated genes were available to regulate ECM − receptor interaction, and the PI3K − Akt signaling pathway et al. (Fig. 6H). These results suggested that there may be a difference in molecular regulation between CSCLPI subgroups.
Impact of tumor microenvironment on biological signatures between CSCLPI subgroupsAt first, the differences in tumor-infiltrating associated cells between CSCLPI subgroups were assessed using ESTIMATE scores. The findings indicated that the high-CSCLPI group had a higher stromal score than the low-CSCLPI category, but there were no differences in immune score between the subgroups (Fig. 7A). We analyzed the types of immune cells infiltrating through ssGSEA. It’s surprising that there was no significant difference in the infiltration of other immune cells between the CSCLPI subgroups, aside from macrophages and regulatory T cells in the high CSCLPI group were higher (Fig. 7B). The immune-related pathways in the high CSCLPI group included cell chemokine receptor (CCR), MHC class I, para-inflammation, and Type I IFN Response, while in the low CSCLPI group, Type II IFN Response was found to be involved in immune regulation (Fig. 7C). TIDE can be used to predict the response to immunotherapy, and T cell dysfunction score can be used to evaluate the function of T cells. The T cell dysfunction score in the high CSCLPI group was found to be higher (Fig. 7D). However, there was no distinction in TIDE score between CSCLPI subgroups (Supplementary Fig. 6A). These findings indicated that immunosuppression was present in the high CSCLPI group, which was one of the causes of poor prognosis in patients with BLCA. There may be no significant difference in the efficacy of immunotherapy between CSCLPI subgroups. Furthermore, we found out that the high CSCLPI group had a higher CAF score than the low CSCLPI group (Fig. 7D). The EPIC analysis further showed that the high CSCLPI group had a more significant CAF score (Fig. 7E). Thus, stromal cells (mainly CAFs) may regulate the differences in biological signatures between CSCLPI subgroups.
Fig. 7
Tumor microenvironment analysis. (A) ESTIMATE analysis between CSCLPI subgroups. The differences in stromal (left) and immune scores (right). (B-C) The immune cell infiltration and immune-related pathways made distinctions between CSCLPI subgroups. (D) TIDE analysis between CSCLPI subgroups. The differences in the dysfunction score (left) and CAFs score (right). (E) The differences in CAFs score between CSCLPI subgroups were calculated using EPIC analysis. (F) CSCLPI subgroups have distinct expression of cancer-associated fibroblast-specific markers. (G) A consensus clustering analysis was conducted on BLCA samples using markers specific to cancer-associated fibroblasts. The optimal groups were determined to be two. (H) The tNES analysis was based on F1 and F2. (I) The difference in CSCLPI score between F1 and F2. (J) F2 had a higher number of patients in the high CSCLPI group
NS: not significantly. *: P < 0.05; **: P < 0.01; ***: P < 0.001; ****: P < 0.0001
The expression of cancer-associated fibroblast-specific markers in BLCA was analyzed. These markers were found to be significantly expressed in the high CSCLPI group (Fig. 7F). Using consensus clustering analysis, TCGA samples were grouped according to cancer-associated fibroblast-specific markers. The best number of groups was two (Fig. 7G, Supplementary Fig. 6B), referred to as fibroblast group 1 (F1) and fibroblast group 2 (F2). The tNES analysis indicated that grouping had an excellent performance in F1 and F2 (Fig. 7H). By comparing the CSCLPI between F1 and F2, we found that F2 had a higher CSCLPI score (Fig. 7I), and the patients in the high CSCLPI group were more present in F2 (Fig. 7J). The findings indicated that the CAFs were responsible for some of the differences in biological signatures between CSCLPI subgroups in BLCA.
Construction and testing of a nomogramClinical characteristics were crucial in determining prognosis. The heatmap showed the relationship between five key genes and clinical characteristics (Fig. 8A). CSCLPI, age, gender, tumor stage, T stage, and N stage were evaluated in univariate and multivariate Cox regression analyses in order to uncover independent risk factors that influence prognosis (Fig. 8B and C). The multivariate Cox regression analysis showed that CSCLPI (HR:3.4305, 95%CI: 1.5896 − 7.4037, P = 0.0017) and N stage (HR: 2.3804, 95%CI: 1.1237 − 5.0429, P = 0.0236) were important risk factors for the prognosis of patients with BLCA. CSCLPI, age, gender, tumor stage, T stage, and N stage were the foundations of the nomogram construction, which was convenient to comprehensively evaluate the 1-year, 2-year, 3-year and 5-year prognosis of patients with BLCA (Fig. 8D). The calibration curves and DCA curves for the nomogram of 1-year, 2-year, 3-year and 5-year prognostic prediction were depicted (Fig. 8E and F). Furthermore, the C-index of the nomogram was 0.743 (95% CI: 0.665–0.821). According to the results, the nomogram was able to predict accurately.
Fig. 8
Construction of a predictive nomogram. (A) The heatmap showed the relationship between five key genes and clinical characteristics. (B-C) Univariate and multivariate Cox regression analyses results of age, gender, tumor stage, T stage, N stage, and CSCLPI. (D) A nomogram for predicting the prognosis of 1 year, 2, 3, and 5 years for patients with BLCA. The prediction of the second patient in the TCGA cohort was depicted. (E-F) The calibration curves and DCA curves of 1-, 2-, 3- and 5-year prognostic prediction for the nomogram. For calibration curves, the more the blue solid line and the black dotted line coincide, the more accurate the prediction was. For DCA curves, if the curve exceeded the horizontal and vertical axes, it implied that the model had certain clinical predictive utility
留言 (0)