
記住我


The RespiRare network allows to collect chILD data on phenotype and investigations with a high degree of precision. The current study included the highest number of chILD ever reported in a single country and retrospectively and prospectively evaluates the prevalence and incidence of chILD as well as the diagnosis repartition, investigations results, treatment and prognosis.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICYIntroductionChildhood interstitial lung disease (chILD) is a heterogeneous group of rare lung diseases with common radiological features, namely interstitial signs on chest X-ray or chest CT scan.1 chILD can present throughout life, from birth to adolescence, with wide variations in severity from pauci-symptomatic diseases to end-stage pulmonary fibrosis leading to pulmonary transplantation or death. chILD classifications usually distinguish two age groups: below 2 years and 2–18 years. One of the first classifications was proposed by Deutsch et al and the most commonly used classification is one of the American Thoracic Society (ATS) classification proposed by Kurland et al in 2013 and the one proposed by Griese et al in Europe in 2018.2–7
Based on these classifications, a limited number of patient series of chILD patients have been reported worldwide (table 1) by single centres or large networks.3 8–20 The reported incidence varied from 1.3 cases/million of children per year in 2009 to 8.2 cases/million of children per year in 2022 and the prevalence from 1.5 cases/million of children in 2017 to 46.5/cases million of children, increasing in recent years, probably because of an improvement of the reporting systems.9 14 15 In France, a first epidemiological study conducted as part of the Reference Centre for Rare Lung Diseases (RespiRare) in 2012 reported 205 patients over 4 years.12
Table 1Childhood interstitial lung disease reported series
During the past 10 years, the RespiRare network has improved its organisation and case collection. In parallel, a national database for rare diseases has been set up in 2019 (BNDMR, Banque nationale de données maladies rares). Thus, the aim of the present study is to describe the epidemiology of chILD in France in terms of prevalence, incidence, respective frequencies of the different aetiologies of ILD, age of onset and mortality.
MethodsPatientsThe study was observational, nationwide, with a retrospective inclusion of the patients between 2000 and February 2022 and a prospective inclusion of the new cases between February 2022 and February 2023. This study was a multicentre study in all the RespiRare centres in France. Patients with ILD born between 2000 and 2022 and aged under 18 years old at diagnosis, were included. The definition of ILD was in line with the ATS Clinical Practice Guideline.2 The aetiology of the ILD was retrieved from the patient’s medical record and was defined by the clinical team in charge of the patient or confirmed by the national chILD multidisciplinary team meeting (MDTm) for those who were alive after the MDTm were implemented in April 2018.21 Patients with bronchopulmonary dysplasia as well as ILD related to infections secondary to primary immune deficiencies were excluded. However, some diseases that include some dysimmune features, such as auto-inflammatory disorders, remain included in the present study. Additionally, in line with other chILD reports, post-infectious disorders manifesting as ILD such as post-infectious obliterans bronchiolitis with no underlying primary immune deficiencies were not excluded.2 3 8 9 14 16–19
Data collectionTo ensure exhaustiveness, the patients were identified in a number of ways: through the national online database for rare lung diseases e-Respirare, through patient files discussed at national MDTm for chILD since April 2018. Additionally, direct secured emails were sent to the clinicians of each RespiRare centre to collect unreported cases.12 21
In order to prospectively evaluate chILD’s incidence, and as recently reported in Spain, the clinicians were contacted again in February 2023, 1 year after the study onset, to collect the data of patients newly diagnosed over 1 year.14 In parallel, an extraction from the official reporting tool for rare diseases, the BNDMR, was performed.
The following data were collected: age at diagnosis, aetiology of chILD, thoracic CT scans, echocardiography with indirect measurement of pulmonary pressures (a pulmonary hypertension defined as a mean pulmonary arterial pressure >20 mm Hg was suspected by an elevated systolic arterial pressure estimated by a systolic tricuspid regurgitation velocity over 2.5 m/s), genetic analyses, bronchoalveolar lavage (BAL) fluid analysis, histopathological findings, treatments and patient outcome at last follow-up (death, alive or lost to follow-up). Because the study spanned over more than 20 years, genetic analyses were heterogeneous over time. A targeted Sanger sequencing was first proposed when there was a high suspicion of an involved gene, and more largely before 2015; a next generation sequencing (NGS) was first proposed in most cases since then, and an exome sequencing was proposed as a second-intention analysis. The composition of the NGS panels is available as online supplemental table S1. The chILD aetiologies were classified into three main groups: disorders more prevalent in infancy, disorder not specific to infancy and unclassified disorders.2
Statistical analysesThe epidemiological information of the overall French paediatric population was extracted from the National Institute for Demographic Studies (Institut National de la Statistique et des Etudes Economiques). The prevalence of chILD in 2022 (number of patients alive with ILD in 2022/number of children alive in France in 2022) and the incidence of chILD between February 2022 and February 2023 (number of new patients with ILD between February 2022 and February 2023/ number of children alive in France in 2022) were calculated. Prevalence and incidence 95% CI were added using the Poisson distribution.
Qualitative/categorical variables are presented as numbers and percentages. Quantitative/continuous variables are presented as median and IQR. χ2 test was used to compare proportions using Biostat TGV software. For small samples (<5), Fisher’s test was used. A p value <0.05 was considered significant. Survival analysis was made using a Kaplan-Meier method and log-rank test on GraphPad Prism software.
ResultsAll 42 RespiRare centres participated in the study. A total of 879 patients with chILD were reported. Of these, 790 patients were included: 78 patients did not meet the inclusion criteria and 11 patients with bronchopulmonary dysplasia were excluded (figures 1 and 2). In parallel, 473 patients with a chILD diagnosis were retrieved from the BNDMR. This register exists since 2019 and is anonymised, thus, it cannot be ascertained that all of these patients were actually included in the 790 patients of the present RespiRare study. As a comparison, within the 2019–2022 period, 277 patients were included in the RespiRare study.
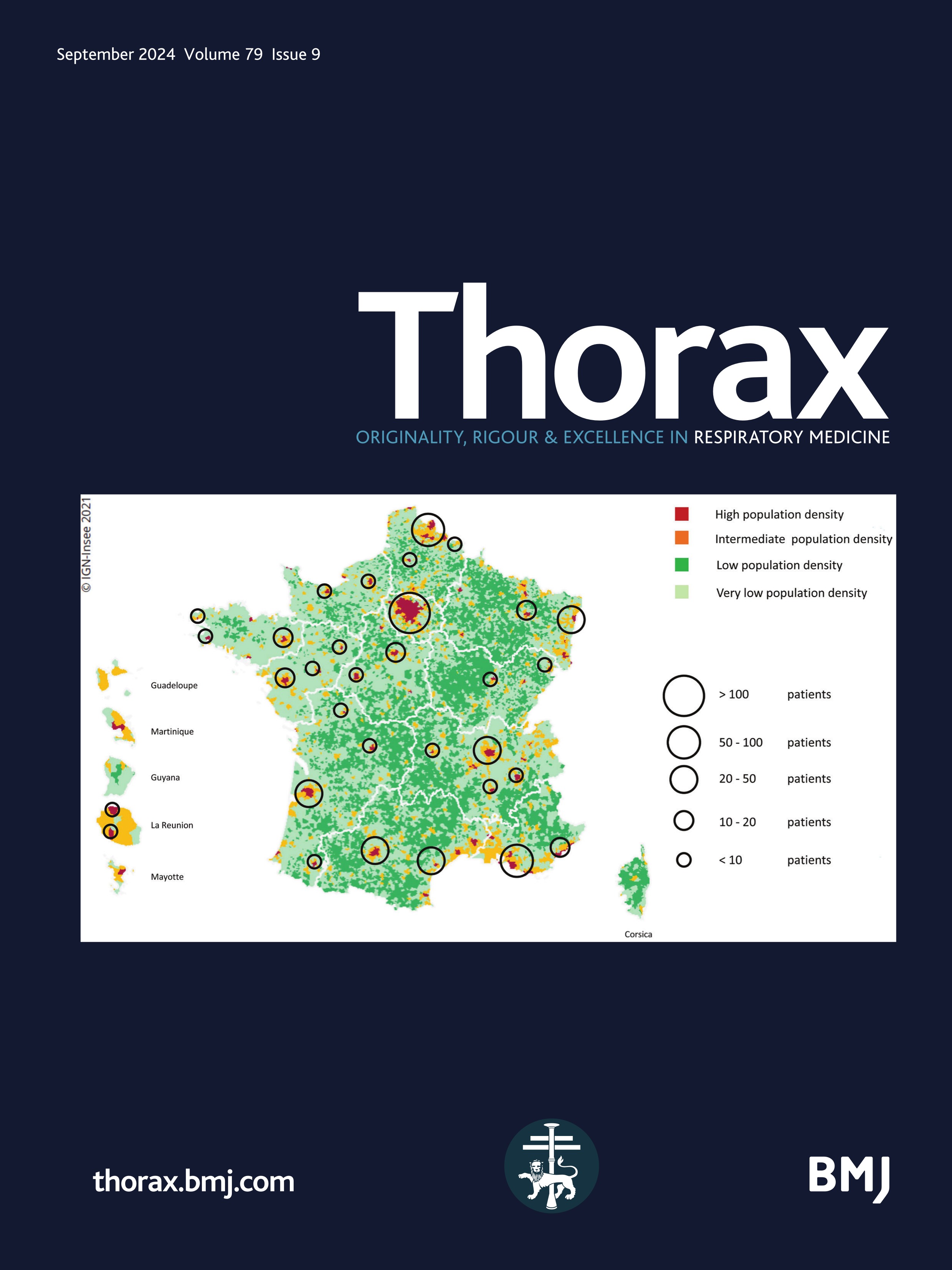
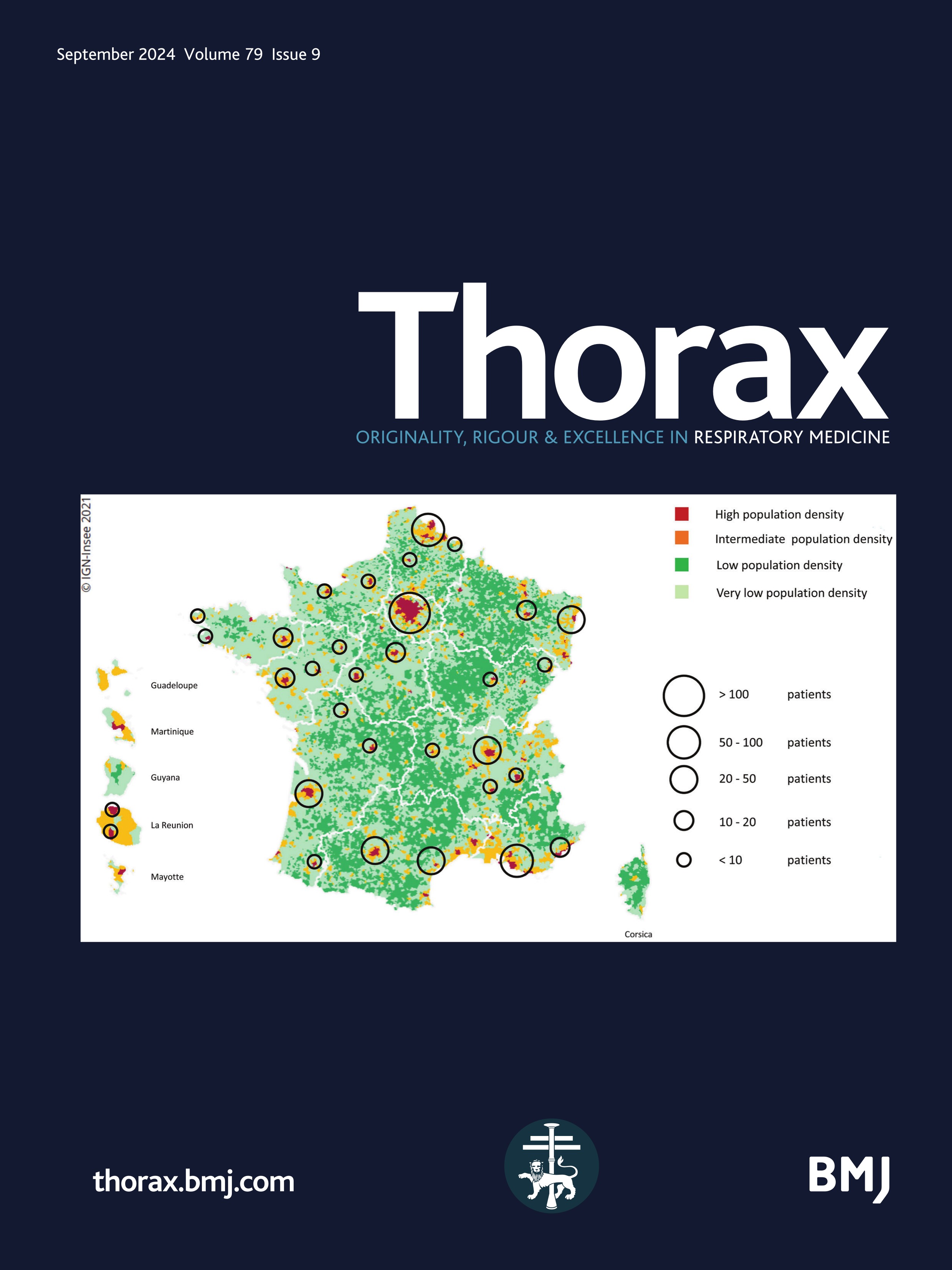
 Figure 1
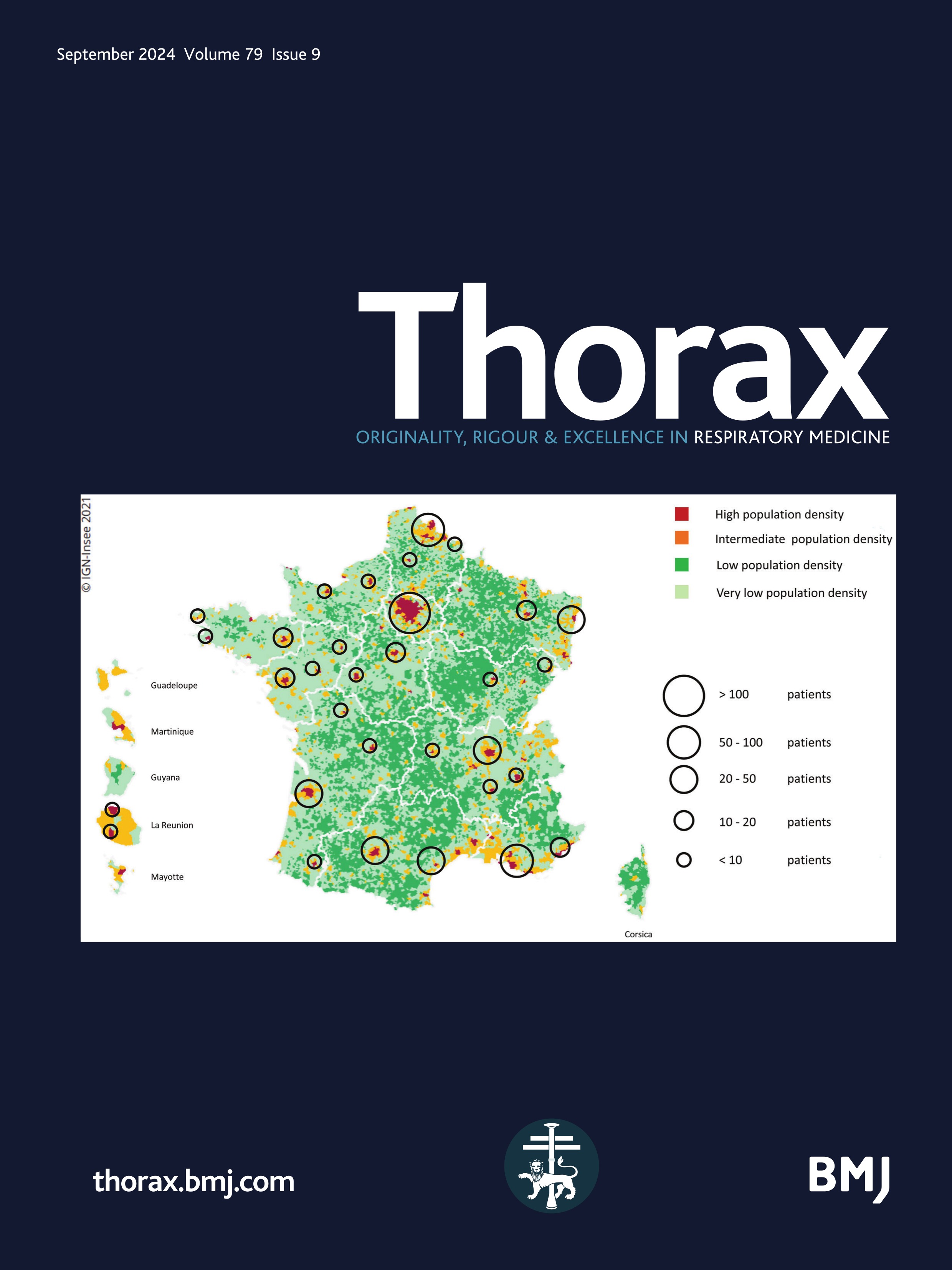
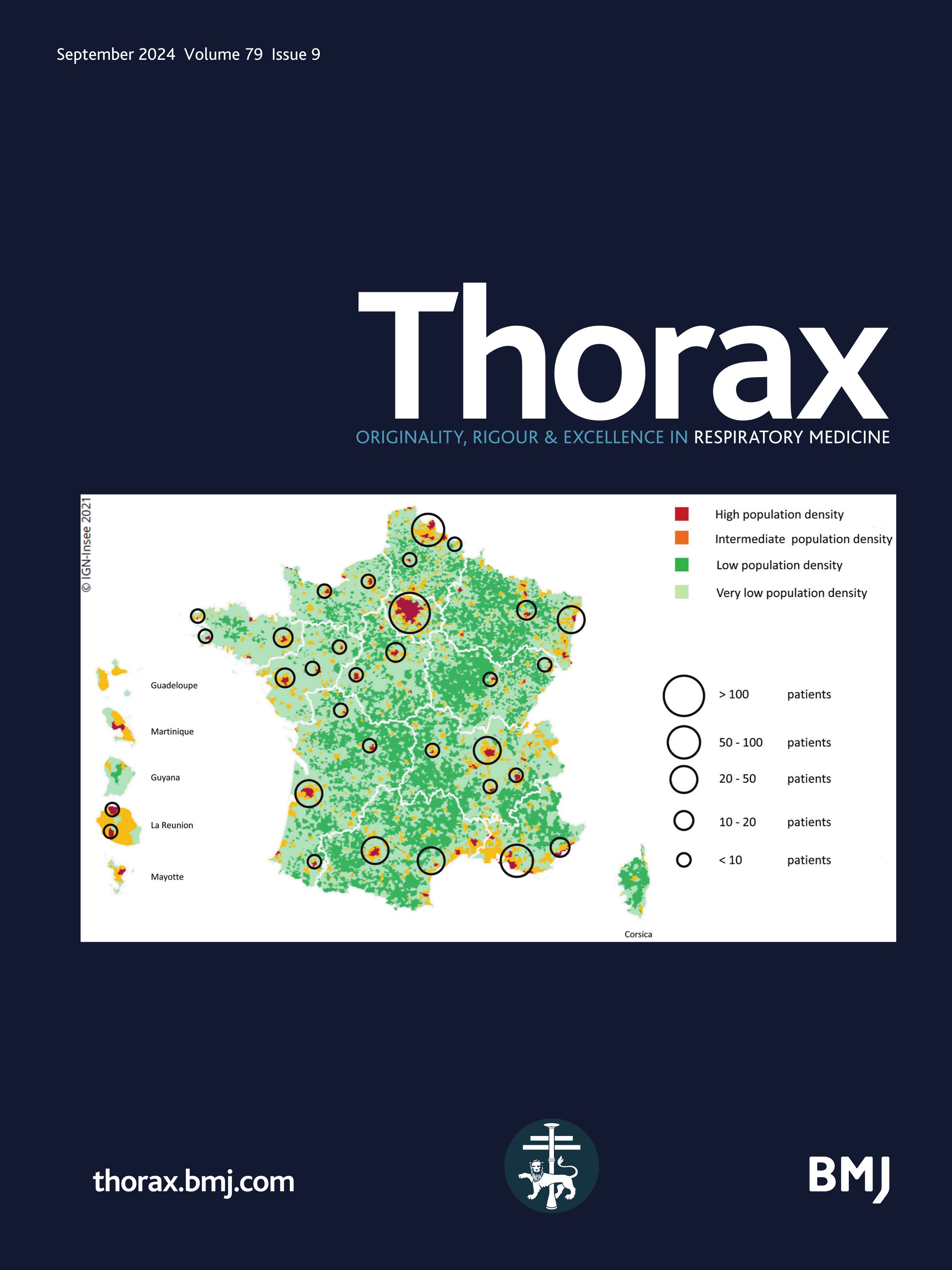
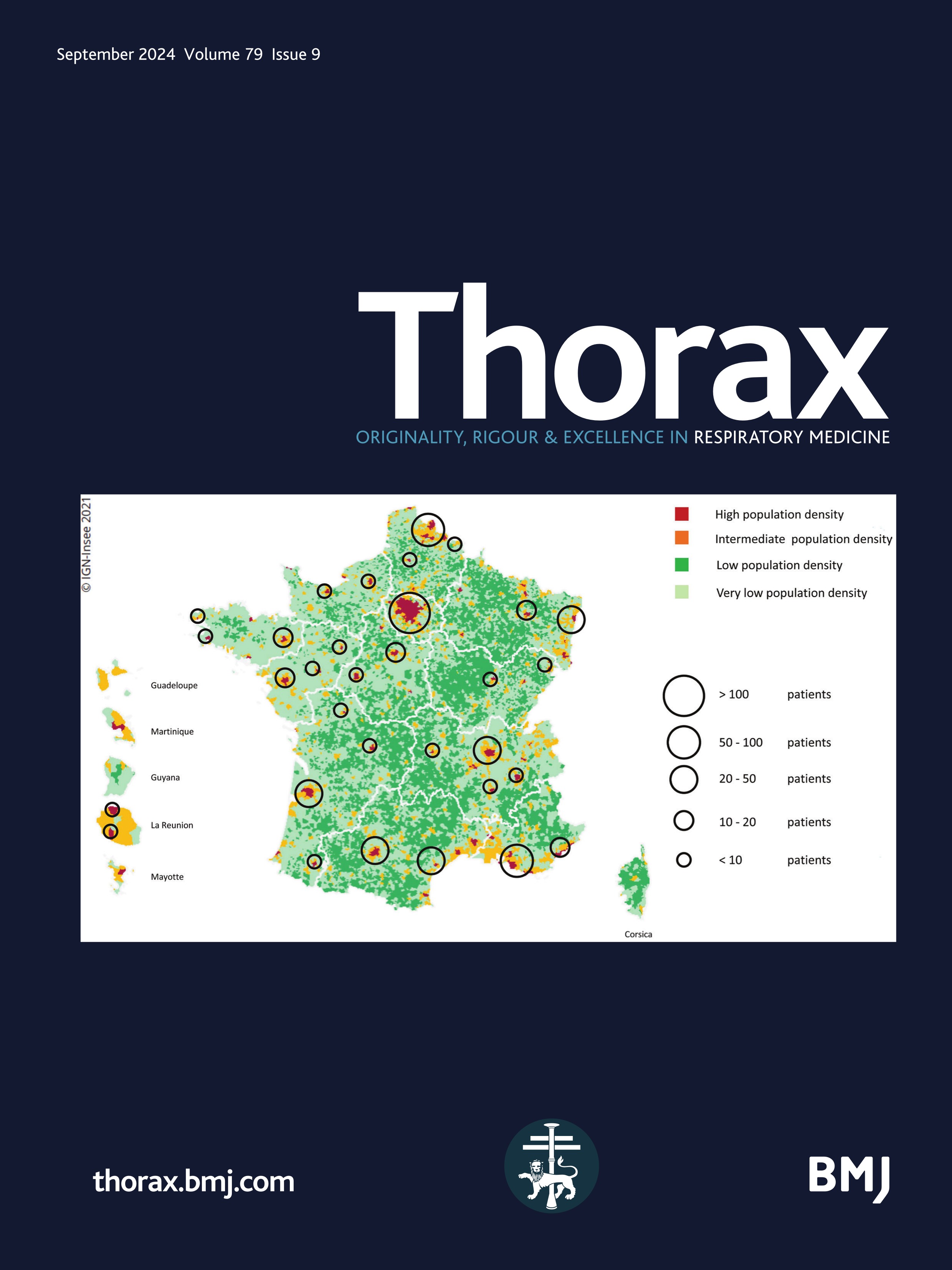
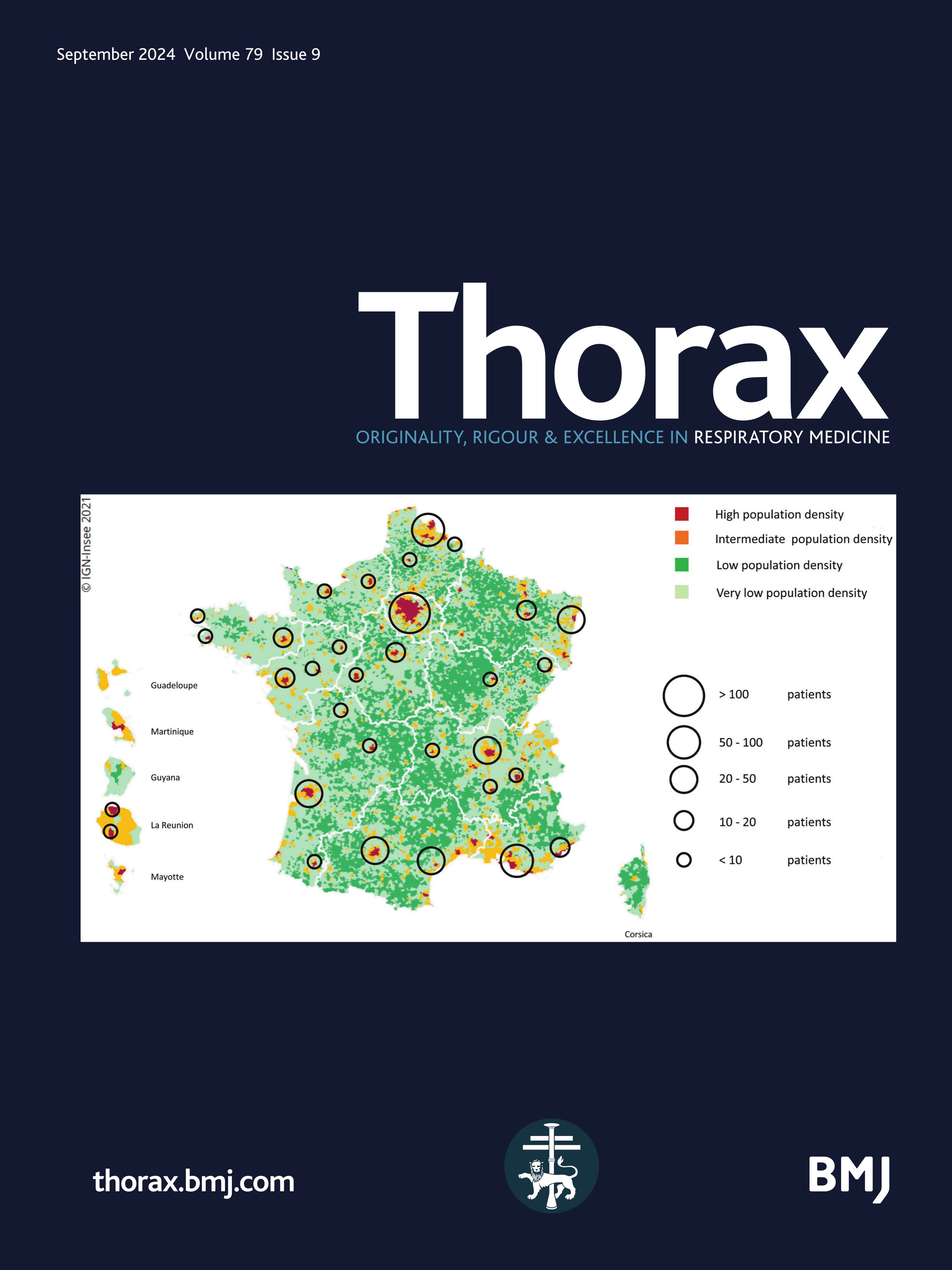
Figure 1 Mapping of the included patients. The RespiRare and collaborating centres who included patients are represented. The larger the circle, the more included patients. The map reflects the population density in France (the highest density of population is represented in red, followed by yellow, light green and dark green). Source: National Institute of Statistics and Economics 2020. Paris area is represented by a single circle (Armand Trousseau hospital n=156, Necker Enfants Malades hospital n=106, Centre Hospitalier Intercommunal de Créteil n=53, Robert Debré hospital n=33, Bichat hospital n=8, Bicètre hospital n=7, Port Royal Hospital n=4, Béclère hospital n=3, Jean Verdier hospital n=1, Saint Denis Hospital n=1), Amiens n=2, Angers n=3, Besançon n=9, Bordeaux n=38, Brest n=4, Caen n=7, Clermont Ferrand n=2, Dijon n=2, Grenoble n=9, La Réunion Island n=15, Lille n=62, Le Mans n=5, Limoges n=2, Lyon n=46, Marseille n=50, Montpellier n=28, Nancy n=10, Nantes n=11, Nice n=20, Nouvelle Calédonie Island n=1, Pau n=1, Polynesia Island n=1, Poitiers n=2, Quimper n=1, Reims n=6, Rennes n=11, Rouen n=4, Saint-Etienne n=1, Strasbourg n=20, Toulouse n=32, Tours n=13.
 Figure 2
Figure 2 Flow chart. After exclusion of 78 patients who did not meet the inclusion criteria (born before 2000 or without available data) and 11 patients with bronchopulmonary dysplasia, 790 patients were included. ILD, interstitial lung disease.
The following results refer to the number of available data. Demographic information showed the majority of patients were full term (528/642 patients, 82.2%). The median age at diagnosis of ILD was 3 months, IQR (0 years–3.5 years). The sex ratio was 1. A familial ILD was reported in 120/710 (16.9%) of the cases. In February 2022, 665 patients were living with chILD in France. Based on a total number of 15 098 384 alive children in France at the same period, the prevalence of chILD was found to be 44 per millions of children (95% CI 40.76 to 47.46). Between February 2022 and February 2023, 67 new patients with ILD were included, leading to a 2022 chILD computed incidence of 4.4 per millions of children (95% CI 3.44 to 5.56).
chILD aetiologies were classified according to diseases group and the age group (<2 years and 2–18 years) (table 2) and (figure 3). Among all 790 patients, the most frequent diagnoses were inherited surfactant metabolism disorders due to surfactant related genes mutations (n=99, 12.5%), neuroendocrine cell hyperplasia of infancy (NEHI) (n=66, 8.4%), alveolar haemorrhage and pulmonary hemosiderosis (n=49, 6.2%), infectious and postinfectious processes (n=46, 5.8%), connective tissue disease-associated ILD: dermatomyositis, systemic lupus erythematosus, scleroderma, idiopathic juvenile arthritis, rheumatoid arthritis (n=30, 3.8%), vasculitis (n=23, 2.9%), sarcoidosis (n=23, 2.9%). Finally, the cause of ILD remained unknown in 238/790 patients (30.1%).
Table 2Type of ILD according to age
 Figure 3
Figure 3 Main aetiological diagnoses of childhood interstitial lung diseases according to age at diagnosis. Number of patients are represented in the different diagnoses according to age at diagnosis (<2 years and 2–18 years).
In the 507/790 patients (64.2%) diagnosed before 2 years, the most frequent diagnoses were surfactant metabolism disorders in 83 patients (16.3%) and NEHI in 60 patients (11.8%).
Among the 228/790 patients (28.9%) diagnosed between 2 and 18 years, the most frequent aetiological diagnoses were alveolar haemorrhage and pulmonary hemosiderosis (n=28, 12.2%), connective tissue diseases-associated ILD (n=26, 11.4%), hypersensitivity pneumonitis (n=20, 8.8%), sarcoidosis (n=20, 8.8%) and vasculitis (n=16, 7%).
A number of investigations were performed during the ILD workup and are reported in table 3: chest CT scan was performed for almost all the patients (644/711, 90.6%) and showed various patterns, ground glass opacities (GGO) being the most frequent in 391/644 (60.7%) (table 4). The remaining patients were either too unstable to undergo a CT scan (67 patients) or had no available CT scan results (79 patients) despite it was performed. For the patients with no CT scan, the diagnosis of chILD was ascertained by a lung sample analysis in 16 patients and based on the chest radiography only in 51 patients. Genetic analysis was performed in 543/706 patients (76.9%), including targeted Sanger sequencing, NGS panels and exomes, and was positive in 227/543 patients (41.8%), mainly identifying a heterozygous or homozygous surfactant-related gene mutation 120/543 (22.1%), a mutation in a gene involved in pulmonary alveolar proteinosis (PAP) or in auto-inflammatory disorders. After a complete work-up, 161/660 patients (24.4%) underwent a lung biopsy of which 113/161 (70.2%) were before the 2015 European recommendations and 48/161 (29.8%) after 2015. Altogether, 191 patients out of 335 patients alive in 2018–2022 (57%) have been discussed at MDTms since 2018.
Table 3Investigations performed during the diagnostic workout
Table 4Main lesions among 644 analysed thoracic CT scans
As obliterans bronchiolitis is not always considered as ILD, depending on the classifications, online supplemental table S2 provides diagnoses of ILD according to the age, with exclusion of 46 patients with postinfectious obliterans bronchiolitis and 85 patients with no available CT scan (not performed or data unavailable) and no genetic or histology ascertaining the ILD. Among these 85 patients, it is important to notice that available data ascertained a cause of ILD for another 16 patients (2 patients with positive hypersensitivity pneumonitis serology, 1 patient with eosinophilic BAL fluid, 13 patients with systemic diseases). In total, for 69 patients, chILD was ascertained only on chest X-ray and history data.
Most patients received corticosteroids (612/790 patients, 77.5%), administrated as intravenous methylprednisolone pulses and/or oral corticosteroids. Other non-specific treatments included azithromycin, hydroxychloroquine, various immunosuppressive drugs. Interestingly, targeted therapies such as antifibrosing therapies, biotherapies, Jak-inhibitors, methionine and whole lung lavages were also used in specific cases (table 5). Oxygen therapy and enteral nutrition were required in 346/665 patients (52%) and 173/642 (27%) patients (nasogastric tube (50%) or gastrostomy (50%)), respectively. A parenteral nutrition was associated with an enteral nutrition in 17/642 patients (2.7%), and was used alone in 4/643 patients (0.6%). Nine (1.7%) patients aged 2.5–17 years benefited from a lung transplantation (table 6).22
Table 5Specific and non-specific therapies used in chILD
Table 6Patients with chILD who received a lung transplantation
Clinical outcome at last follow-up was available in 705/790 patients (89.2%) (table 7). A total of 580/705 patients (82.3%) were alive at last follow-up with a median age of 7 years, IQR (2.6 years–13.7 years). The 5-year survival rate was 57.3% for patients diagnosed before 2 years and 86% for those diagnosed between 2 and 18 years.
Table 7Evolution at the last follow-up
Finally, 125/705 patients (17.7%) died at the median age of 6 months, IQR (0.1 years–2.9 years) including two patients after lung transplantation. The Kaplan-Meier curve according to age at diagnosis is provided in figure 4 and showed a reduced survival of the patients who developed chILD before 2 years (log-rank p<0.0001 and log-rank for trend p=0.016).
 Figure 4
Figure 4 Kaplan-Meier survival curve according to age at diagnosis. Survival rate (%) at the last follow-up (years) according to age at diagnosis (<2 years and 2–18 years). A log-rank test assessed the differences in survival rates.
DiscussionIn this largest epidemiological study in chILD in a single country over 23 years, we assessed a prevalence of chILD in 2022 of 44 cases per millions of children and an incidence of chILD between February 2022 and February 2023 of 4.4 cases per millions of children in France. These numbers are much higher than in previous epidemiological studies and similar to the latest Spanish study (table 1).3 8–19
Consistently with previous studies, most patients were diagnosed at birth or shortly after birth with a median age at diagnosis of 3 months, IQR (0 year–3.5 years). Below 2 years of age, the most frequent chILD aetiologies were surfactant metabolism disorders (16.3%) and NEHI (11.8%). Between the ages of 2 and 18 years, the most frequent aetiological diagnoses were diffuse alveolar haemorrhage (12.2%), connective tissue diseases-associated ILD (11.4%), hypersensitivity pneumonitis (8.8%), sarcoidosis (8.8%) and vasculitis (7%). In comparison, the 2007 US study based on histological analyses reported fewer surfactant dysfunction disorders (10.9%), a similar number of NEHI and pulmonary glycogenosis (14.5%) and more lung growth abnormalities reflecting impaired alveolarisation (27.9%) in infants. In older patients, the authors found mainly opportunistic infection in immunocompromised host (12.1%) and infections/postinfectious process (10.3%).6 However, these discrepancies could hardly be compared as this study was based on histological analyses and included immunocompromised host aetiologies. The methodology of the present study was chosen to allow a comparison with the recent Spanish study and found a comparable distribution of the diagnoses, increasing the reliability of both recruitment strategies.
Despite some patients with chILD could have been missed, especially those who presented at birth with a rapid fatal outcome such as developmental disorders or surfactant metabolism disorders, it can be supposed that the original use of three different methods of data collection (e-RespiRare, direct contact with clinicians, reports from MDTms), allowed this study to achieve good coverage. This study gathered the information from 42 centres in France enabling data to be collected from a larger number of patients than is reported at European level with non-systematic reporting systems. As France pioneered the collection in rare lung diseases data collection with the launch of the RespiRare network in 2008, a large amount of data was collected for each patient, and a follow-up evaluation was provided.12 Interestingly, it can be observed in figure 1 that the rate of chILD per centre is consistent with the population density, highlighting a homogeneous ability to diagnose chILD and to collect the data over the national territory. Of note, the national platform for reporting rare diseases (BNDMR) only retrieved 473 chILD since 2019. This system allows to declare cases and to collect a minimum set of data for patients with rare diseases using an ORPHA code. Following informed consent, this register can collect data directly or by systematic transfer from the patient’s medical form in a growing number of centres. Although not exhaustive to date, this system will probably be of great benefit in increasing collection of data in rare diseases.
The diagnostic workup showed that genetic testing was used much more frequently than lung biopsy and that it enabled allowing a definitive diagnosis in a greater number of cases (41.8%).23 Mutations in surfactant-related genes were the most commonly reported aetiologies (22.1%), but rarer molecular disorders have also been diagnosed using various NGS panels (67% of the positive results were found through chILD NGS panel, see online supplemental table S1) or whole genome sequencing (table 3). This observation confirms that the indications for lung biopsy are currently decreasing while new biological tests such as the interferon signature, or new molecular techniques are increasing.24 25 Due to constant improvement of genetic screenings and molecular knowledge of chILD, it seems crucial to store DNA and/or any possible lung tissue material for every patient with chILD or suspicion of chILD without a definitive diagnosis. A molecular analysis should be performed in all cases of chILD, with a whole exome or genome sequencing if first-intention NGS panel is negative. This genetic diagnostic workup has been set-up since 2021 in France where all patients without an identified cause of rare disease through NGS panels can benefit from a whole genome analysis (Plan France Médecine Génomique https://pfmg2025.aviesan.fr/).
This study also highlighted an intriguing finding related to the high rate of positive Pneumocystis jirovecii in chILD patients (42 patients out the 196 researches, 21.4%). Interestingly, the largest group, 13 patients, had an NEHI. At this stage the rational for such observation is not clear but as NEHI pathophysiology remains unclear, a potential role of P. jirovecii colonisation needs to be further investigated.
As chILD diagnostic workup remains a long journey, the stepwise approach to an aetiology is increasingly discussed at a national level during the monthly chILD MDTms organised by the RespiFIL network.21 26 In adults, such MDTms are even considered as the gold standard for ILD diagnosis.26 27 In children, these meetings has been widely used in the reported population and highlighted the close collaborations that are established between the clinicians, radiologists, geneticists and pathologists involved in chILD.21 26 28 These meetings are now well organised and easily accessible to all clinicians in France and abroad (www.respiFIL.fr). They are intending to be launched in English language at a European level within the ERS Clinical Research Collaboration (CRC) for chILD (CRC-chILDEU) and the European Reference Networks (ERN)-lung Clinical Patient Management System.
This study confirms that chILD is associated with a high morbidity and disease burden.29 Half of the patients required oxygen therapy and 27% required enteral nutrition. Most patients received multiple oral or intravenous treatments: corticosteroids were largely used, as well as azithromycin, immunosuppressive drugs and hydroxychloroquine. Their use is mostly based on expert advices while clinical trials are very few owing to high costs and small numbers of included patients.30–32 To date, very few patients have benefited from targeted treatments, but their number may increase in the coming years with the development of international or even national clinical trials (nintedanib for fibrosing ILD, methionine for MARS1-related PAP, respectively).33–35 The InPedILD trial is a promising and innovative example that is currently testing nintedanib, which efficacy has been proven in adults, using appropriate dosages in children. Retargeted drugs may also be part of the future chILD treatments of chILD as recently shown with JAK-inhibitors for auto-inflammatory diseases or CFTR modulators for ABCA3 mutations.36–38 Even being an optimistic perspective in such rare diseases, large and well-defined patient cohorts are instrumental to the development of such clinical trials. In the meantime, as recently pointed out in chILD, in vitro functional studies of new targeted drugs are probably a more realistic option to be developed.38
Therapeutic optimisation and identification of prognosis factors are also required, as prognosis of chILD remains poor with 17.7% of premature death at a median age of 6 months (0.08 years–2.9 years). Very few prospective evaluations of chILD are available. Recently, Cunningham et al provided a 1-year prospective evaluation of 127 young patients (median age 0.9 (IQR 0.3–7.9)) with chILD and found as deleterious prognosis factors an age below 6 months, a SpO2 <94% and developmental/surfactant disorders (this last group accounting for 20 patients).39 In line with these results, the present study, the 5-year survival was 57.3% for patients diagnosed before 2 years of age and 86% for patients diagnosed between 2 and 18 years of age, reflecting the poor prognosis of congenital disorders such as diffuse developmental disorders or surfactant metabolism disorders, in particular bi-allelic mutations of SFTPB and ABCA3. Ongoing longitudinal studies will help to confirm these results as, even being large and exhaustive; the median duration of follow-up of this study is limited to 3 years (0.75 years–7 years). The development of transition of care programmes from childhood to adulthood is an interesting perspective for improving the longitudinal quality of the data and assessing patients prognosis over time in terms of survival, development of pulmonary fibrosis, long-term treatment side effects and quality of life.29 40 41
The study displays some limitations linked to its retrospective design, which induces a loss of information for some patients and/or patients lost to follow-up or deceased, especially for neonatal deaths. The expertise of the clinicians of the RespiRare centres reinforces the veracity of their diagnosis. However, the lack of confirmation of the chILD aetiology by the MDTm for a large number of patients still constitute a bias and remains to be improved in the coming years. Another limitation of this study is linked to the ATS classification that was used to compare our results with the 2022 Spanish study.14 This classification is now 10 years old, and some subcategories could be re-evaluated, in the light of newly identified diseases and with a better harmonisation with adults ILD classifications. Moreover, this classification, like most of chILD classifications, includes disorders such as Trisomy 21 related ILD, or diseases of the distal bronchioles such as postinfectious diseases—obliterans bronchiolitis—or NEHI, whereas some other classifications do not, at least for obliterans bronchiolitis. This choice probably originates from the imaging pattern of obliterans bronchiolitis that may appear as GGO revealing ventilation mosaicism more than ILD. In the present study, online supplemental table S2 excludes obliterans bronchiolitis, without major changes in the remaining chILD frequencies.
Finally, chILD is less rare than previously reported, and remains a group of severe diseases with limited therapeutic options. The next challenge will be to take advantage of well-organised international networks, such as the CRC chILD and the ERN-lung to set-up systematic cohorts and registries—at least for certain chILD aetiologies—to develop and test new therapeutic options and to prepare new classifications including adult ILD diagnoses and new guidelines for the management and transition of care of these patients.
Data availability statementData are available upon reasonable request.
Ethics statementsPatient consent for publicationEthics approvalThis study involves human participants and the e-RespiRare database and data collection were approved by the French national data protection authorities: 'Commission Nationale de l’Informatique et des Libertés, CNIL n°08.015bis' and the 'Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé, CCTIRS 20080328'. The present study was approved by the ethic committee of the French pulmonology society (CEPRO) under the number 2022-054. The data collection received the agreement of the General Data Protection Regulation board of the institution under the number 20230201160215. The scientific committee of the BNDMR approved the study (n° D2023-0005). The parent’s consent was obtained before including the data into the e-RespiRare database and/or the MDT reports.
AcknowledgmentsWe thank the Assistance Publique-Hôpitaux de Paris (APHP), Sorbonne Université (SU) Paris, France, and the Institut National de la Santé et de la recherche Médicale (Inserm). We thank the national networks for rare lung diseases: Centre de référence des maladies respiratoires rares (RespiRare, www.respirare.fr), the Filière de santé pour les maladies respiratoires rares (RespiFIL, www.respifil.fr), the Rare diseases Cohort project for ILD (RaDiCo-PID) and the ERN-lung. We thank the Banque National de Données Maladies Rares (BNDMR) for their collaboration. The chILD studies are part of the European respiratory Society (ERS) Clinical Research Collaboration for chILD (CRC chILDEU), with the support of the European Lung Foundation (ELF) chILD group.
留言 (0)