
記住我


To interrogate the effect of Aβ on proteostasis we utilized the C. elegans strain, GMC101, which expresses the human, pro-aggregating, and pathogenic Aβ1-42 peptide in muscle tissue, here referred to as simply Aβ [17, 29]. When cultured at 20 °C, animals exhibit Aβ at low levels but at 25 °C they induce elevated levels of Aβ. After 24 h of Aβ induction, > 80% paralyze [29]. We allowed animals to develop at 20 °C, i.e., without Aβ expression, and then moved them to 25 °C at the beginning of adulthood. Animals were then collected after 24 h. We extracted insoluble, aggregated proteins by serial washing lysates with 1% sodium dodecyl sulfate (SDS) buffer. We used protein mass spectrometry and data independent acquisitions (DIA) to identify and quantify proteins, as previously described [28, 30, 31] (Fig. 1A). Background genotype-control animals (CL2122) lacking the Aβ transgene were cultured and processed in parallel for comparison (Fig. 1A).
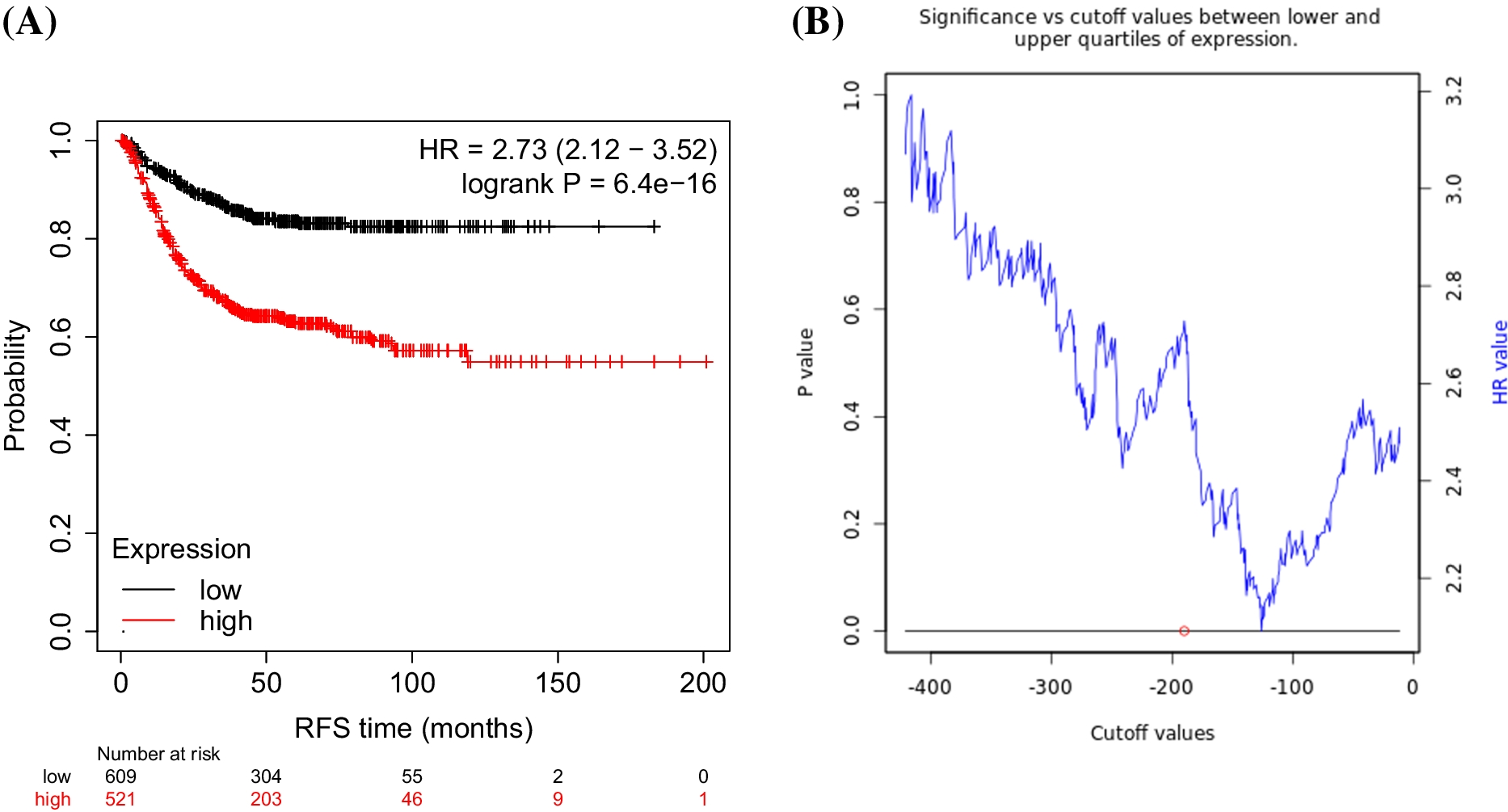
Fig. 1
Aβ drives proteome-wide protein insolubility that resembles normal aging. A Schematic of experimental procedure, created with Biorender.com. B Schematic overview of proteostasis machineries and mitochondrial proteins enriched in the Aβ-driven insoluble proteome. C Total insoluble protein Intensity in worms expressing Aβ vs genotype control strain CL2122, all values normalized to the average of the control in each experiment n = 4, error bars = SEM, Mann-Whitney Test, created with Biorender.com. D Barplot of selected enriched GO terms, KEGG pathways and Wikipathways in the Aβ insoluble proteome, Benjamini Hochberg FDR <5% (see supplementary tables for complete list). E Left: Overlap of proteins enriched in the insoluble proteome of old C. elegans from two publicly available datasets [5, 28] p < 0.0001 Fischer’s exact test. Right: Overlap of aging insoluble proteome with Aβ-driven insoluble proteome, p < 0.0001 Fischer’s exact test. F KEGG disease pathways enrichment of human orthologues of core insoluble proteome, Benjamini Hochberg FDR < 5%
Aβ expression in young adult animals caused a robust, proteome-wide increase in the total amount of insoluble protein, impacting a wide array of essential functions (Fig. 1B, C). We identified and quantified peptides representing 1704 proteins in the insoluble fraction across four biological replicates, of which 593 proteins robustly increased due to Aβ expression across independent experiments (Fig. S1A). Cytoplasmic and organelle-specific heat shock proteins (HSPs) from across all major organelles were enriched in the insoluble fraction (Fig. S1B). Several proteins involved in maintaining proteostasis also became insoluble such as the proteasome regulatory “lid” complex, the TriC chaperonin complex, and key lysosomal proteins (Fig. 1B). Furthermore, we observed an increase in protein insolubility of almost every ribosomal subunit and translation accessory factors (Fig. 1B). Gene Ontology (GO) and pathway enrichment analysis revealed that oxidative phosphorylation (Ox. Phos), ribosomes, the mitochondrial unfolded protein response (mitoUPR), and “Determination of lifespan” were the most enriched terms in the insoluble proteome (Fig. 1D, Supp. Table 2). 42 of the 70 proteins in the C. elegans proteome bearing the annotation “Determination of Lifespan” became insoluble due to Aβ (Fig. 1D). Indeed, the Aβ insoluble proteome was highly enriched with mitochondrial proteins (109/593); particularly those involved in the ETC and TCA cycle (Fig. 1B, D, Supp. Table 1). Consistent with this, the mitochondrial unfolded protein response (mitoUPR) chaperone HSP-6 increased 9.2-fold in the insoluble fraction. Forty-three of the 88 ETC proteins became insoluble due Aβ expression (ETC complexes detailed in Supp. Table 3). Nuclear-encoded ETC complexes, were particularly vulnerable to insolubility (Fig. 1B) whereas mitochondrial DNA-encoded ETC complex proteins were not: only one mtDNA-encoded, complex IV protein became insoluble (Supp. Table 3). The skewed representation of nuclear-encoded subunits could be explained by deficits in the mitochondrial protein import machinery; proteins failing to locate the mitochondria could be subject to aggregation. For example, we observed a significant increase in insolubility of mitochondrial outer membrane proteins responsible for importing nuclear encoded ETC proteins. All major proteins of the TOM complex: TOM-20, TOM-22, TOM-40, and TOM-70 became insoluble (Supp. Table 1). We also identified GOP-3, the orthologue of the outer membrane complex protein SAMM50 [32], which is required for threading of mitochondrial β-barrel proteins into the outer and inner membrane bilayers [33], along with several small molecule transporters: VDAC-1, ANT-1.1 and MTCH-1 (Fig. 1B, Supp. Table 1). Interestingly, single nucleotide polymorphisms (SNPs) in several TOM complex genes have been linked with AD risk, most prominently TOMM40, suggesting an important role for mitochondrial protein import in the etiology of AD [34,35,36] (Fig. 1B).
We questioned if many of the proteins in the Aβ-driven insoluble proteome have been shown to directly interact or co-aggregate with Aβ in the literature. Comparing laser microdissection proteomics data from human AD senile plaques with the Aβ-driven insoluble proteome we found a highly significant overlap [37] (Fig. S1B, Supp. Table 4). Specifically, ~ 1/3 of the proteins which co-aggregate with Aβ in plaques also become insoluble due to Aβ expression in C. elegans, almost all of which are intracellular proteins (Fig. S1B & S1C, Supp. Table 4). Similarly, we find that C. elegans orthologues for 54% (15/28) of the proteins identified to reproducibly interact with short Aβ peptides in transfected HEK293T cells, were enriched in the insoluble proteome after Aβ expression [38] (Supp. Table 4).
The Aβ-driven insoluble proteome is highly similar to the aging-driven insoluble proteomeNormal aging causes a significant increase in the amount of insoluble protein, and we noticed many of the same proteins that typically become insoluble during normal aging were being driven to insolubility by Aβ [4, 5, 9]. This led us to question the extent to which these two insoluble proteomes were similar. To assess this rigorously, we generated a list of proteins that robustly insolubilize during normal aging by overlapping two published aging insoluble proteomes from different laboratories [5, 28]. This resulted in an overlap of 457 proteins that reliably become insoluble during aging (p < 0.0001, Fischer’s exact test) (Fig. 1D). Strikingly, when we compared this with the Aβ-driven insoluble proteome, we found that 305 proteins, or 66%, became insoluble under both conditions (p < 0.0001, Fischer’s exact test) (Fig. 1E). Moreover, when we compared the GO biological processes (BPs) represented in the aging and Aβ insoluble proteomes, we uncovered an 89% overlap, indicating that aging and Aβ drive insolubility of proteins involved in almost identical biological processes (Fig. S1D). By comparison, previous work showed very minimal overlap (< 3%) between the proteins which become insoluble under different stress conditions, such as hsp90 inhibition, proteasome inhibition, and polyglutamine repeat expression, highlighting the remarkable similarity between the aging-driven and Aβ-driven insoluble proteomes [39]. The fact that two-thirds of proteins that become insoluble during normal aging also do so after Aβ expression in young animals suggested to us that we have identified a core set of vulnerable proteins relevant to age-related ND, which we refer to herein as the “core insoluble proteome” (CIP).
We suspected from previous work [4] that the CIP might be particularly enriched with modulators of aging and, indeed, we found that the CIP was highly enriched with modulators of lifespan. Specifically, 100 proteins, or roughly one-third of the CIP, modulate lifespan according to the GenAge database [40]; with 80% of those proteins extending lifespan when their expression is reduced or eliminated (Fig. S1E&F). Furthermore, using published whole-genome RNA interference (RNAi) screens [41,42,43,44,45], we found that knockdown of approximately one in six of the proteins in the CIP has been shown to improve disease pathology across Huntington’s disease (HD) and Parkinson’s disease (PD) proteinopathy models, demonstrating that the CIP is likely enriched with drivers of disease (Supp. Table 5). This led us to directly question if the orthologues of CIP proteins had been implicated in neurodegenerative proteinopathies in humans. To do so, we performed disease pathway enrichment analysis with the human orthologues of the CIP and found a highly significant enrichment for a range of neurodegenerative diseases including prion disease (Pr), AD, PD, and HD (Fig. 1F, Supp. Tables 6 and 7). In almost all cases, the proteins were annotated against all four neurodegenerative diseases, suggesting a common mechanism might underly their associations with disease (Supp. Tables 6 and 7). These data point towards protein insolubility as a common mechanism.
The CIP is enriched with supersaturated but not intrinsically aggregation-prone proteinsWe questioned if the intrinsic properties of the proteins of the CIP proteins might explain the common formation of insoluble aggregates under Aβ and aging. To test this, we applied a series of prediction algorithms to test if the insoluble proteome contained intrinsically aggregation-prone or insolubility-prone proteins and if the proteins in the CIP have higher liquid–liquid phase separation (LLPS) propensity than the whole proteome. We included the following five amyloidogenic, aggregation-prone proteins as “positive” comparators: Aβ precursor protein A4, prion protein, α-synuclein, β-2 microglobulin, and amylin. Surprisingly, using the Zyggregator aggregation prediction method [46], we discovered that the core insoluble proteins were significantly less aggregation-prone than the reference proteome (Fig. 2A). Similarly, using the CamSol solubility prediction score [47] we found that the CIP had a significantly higher median solubility prediction than the reference proteome (Fig. 2B). Conversely the CIP had a higher catGRANULE LLPS propensity score [48] than the reference proteome, suggesting that CIP proteins are prone to form LLPS bodies (Fig. 2C). Taken together, these predictions suggest that protein intrinsic aggregation propensity does not account for the CIPs increase in insolubility, but that LLPS propensity could play a role in their enrichment in the insoluble proteome.
Fig. 2
The core insoluble proteome is enriched with supersaturated but not aggregation-prone proteins. A Violin plot of CamSol intrinsic solubility score distribution for each proteome. B Violin plot of Zyggregator aggregation propensity score distribution for each proteome. C Violin plot of catGRANULE RNA granule prediction score for each proteome. D Violin plot of supersaturation (σf Log10) score distributions for each proteome. In each case, the background insoluble is all proteins identified in the insoluble proteome across any experiment and the whole proteome is the C. elegans reference proteome. Disease amyloids = Aβ precursor protein, ⍺-synuclein, prion protein, β-2 microglobulin, and amylin. For clarity, only comparisons that were statistically significant are shown, all other pair-wise comparisons were not significant to p.adj < 0.05, Kruskal–Wallis test with Dunn’s correction
Supersaturation refers to a state where the concentration of a protein in a solution exceeds its equilibrium solubility. Supersaturated proteins have been shown to be enriched in several neurodegenerative disease KEGG pathways including AD, PD, and HD [49]. Given the overrepresentation of these pathways in the CIP (Fig. 1E), we postulated that the CIP might be particularly enriched with supersaturated proteins. Indeed, we found that the average supersaturation score of the CIP was 94-fold greater than the C. elegans reference proteome [49] (Fig. 2D). This suggests that proteins driven to aggregate by both aging and Aβ are highly supersaturated proteins, which is consistent with previous literature showing that AD plaque proteins are particularly supersaturated [49].
This finding implies that insolubility may be simply driven by expression changes, pushing proteins beyond their solubility limit. Previous groups have shown that age-related protein insolubility does not correlate with changes in protein abundance [50]. However, to assess this in the context of the CIP, we sourced publicly available RNA sequencing data and lysate proteomics for the GMC101 Aβ model [51]. We found that 93% of CIP proteins which become insoluble due to Aβ did not change at the mRNA level and 94% did not change at the level of overall abundance after Aβ induction (Fig. S2A) [51]. In parallel, we sourced aging mRNA expression data from the “metaworm” dataset which includes 60 aging transcriptomic profiles for wild-type animals [52]. We performed Spearman’s rank correlation analysis on mRNA expression changes of CIP proteins from day 2 to day 10, matching the age at which animals were collected for insoluble proteomics. Most CIP proteins did not significantly correlate with aging and, of those that did, there was an equal proportion of genes that increased or decreased (Fig. S2A&B). Finally, using publicly available lysate proteomics data for aging wild-type C. elegans, we found that the overall abundance of 94% of CIP proteins did not significantly change during aging (Fig. S2A) [53]. Collectively, these data strongly refute the notion that expression changes are a primary driver of CIP protein insolubility.
Intriguingly, while the aging-driven insoluble proteome was indistinguishable from the CIP, differences emerged between the CIP and the Aβ-driven insoluble proteome (Fig. S2B-E). Specifically, the Aβ-driven insoluble proteome displays greater aggregation propensity and lower solubility than the CIP (Fig. S2B & C) [49]. These data are consistent with the notion that Aβ and aging drive protein insolubility through a common mechanism but that Aβ may also cause insolubility through a distinct mechanism, likely by direct interaction and seeding of aggregation. Together these findings support the hypothesis that aging and Aβ act synergistically in the context of AD to accelerate age-related loss of proteostasis.
Protein insolubility impacts biological processes that increase human age-related disease riskKEGG analysis of the CIP showed that in addition to bearing annotations for neurodegenerative disease pathways, many insoluble proteins bore pathway annotations for non-neurological, age-related diseases, such as non-alcoholic fatty liver disease and diabetic cardiomyopathy (Supp. Tables 9A & B). This led us to question if the relationship between the insoluble proteome and chronic age-related disease (CARD) could go beyond simply neurodegeneration. We reasoned that biological pathway dysfunction due to insolubility could be an underappreciated driver of CARD risk. Previous GWAS analyses demonstrated that common biological processes underly a whole array of diverse CARDs [54]. We therefore applied this analytical pipeline to query the relevance of protein insolubility and biological processes implicated in CARD risk (Fig. 3A). Specifically, we compared biological processes impacted by insolubility against those implicated in the risk of developing 38 distinct CARDs (Supp. Table 10). We found that, on average, proteins in the CIP shared biological processes with 18 of the 38 different CARDs (Fig. 3B). Moreover, 88% of the CIP proteins shared annotations with diseases spanning four or more of five broad CARD categories: neurodegenerative, metabolic, cancer, cardiovascular, and “other” (capturing disparate CARDs such as macular degeneration, rheumatoid arthritis, and osteoporosis) (Fig. 3C). This association was enriched compared with the experimental background insoluble proteome (i.e., all proteins that could be identified in the insoluble fraction across all experimental conditions) and highly enriched compared with proteins in the whole proteome for which the average protein had no shared processes with CARDs (Fig. 3B).
Fig. 3
The insoluble proteome is enriched with modifiers of age-related disease risk. The core insoluble proteome is enriched with modulators of age-related disease risk. A Schematic representation of GWAS data53 used to compare GO biological processes (BP) shared across chronic, age-related diseases (CARDs) with the BPs represented in the CIP, created with Biorender.com. B Distribution of the number of CARDs that share biological processes with proteins found in CIP, the background insoluble proteome, and the C. elegans reference proteome with the median value displayed, Kruskal–Wallis test with Dunn’s, error bars = range. C Frequency distribution of the number of broad CARD classes that share one or more biological processes with proteins found in the CIP, the insoluble proteome, and the proteome. D Distribution of number of non-age-related diseases, Kruskal–Wallis test with Dunn’s, error bars = range. E Proportion of diseases sharing annotations with the insoluble proteome normalized to the number of diseases tested in either age-related disease or non-age-related disease categories
We questioned if this enrichment was specific for age-related diseases or simply an association with diseases more generally. We therefore repeated the analysis with 12 non-age-related diseases such as schizophrenia, childhood leukemia, and autism. We found that the CIP shared biological processes with only 3 diseases and there was no statistical difference between the CIP and the experimental background insoluble proteome (Fig. 3D). Normalizing by the number of disease comparisons showed a roughly twofold higher enrichment for CARDs over non-age-related diseases (Fig. 3E). Taken together these data suggest that the CIP is enriched with processes whose dysfunction contributes to disease risk but age-related disease risk in particular.
It is worth noting that TOMM40, which we identified in the Aβ-driven insoluble proteome and two independent aging-driven insoluble proteome datasets [10, 28], is one of only a very small fraction of SNPs (2.5%) that increase disease risk for > 3 broad categories of CARD suggesting that TOMM40 insolubility, and mitochondrial protein import deficits, could be central drivers of age-related disease [54].
The biological processes common to the insoluble proteome and all 5 broad CARD categories included: immune activation and stress response pathways; growth signaling, such as fibroblast growth factor (FGF) and WNT signaling; RNA splicing and regulation of expression; and tissue homeostatic processes important for development and wound healing, such as ECM organization, cell migration and cell differentiation (Fig. S3A). Except for a few rare cases, soluble proteins lose their biological function when they become insoluble, and therefore, these data provide suggestive evidence that insolubility of a core set of vulnerable proteins might promote risk of not only neurodegenerative disease but CARDs more broadly.
The CIP can be used to identify therapeutic targets for Aβ toxicityGiven that one in six of the CIP proteins has been shown to alleviate toxicity in either HD or PD models, we speculated that insoluble proteins in the CIP could be playing a direct role in the toxicity of Aβ, and therefore reducing their expression may be beneficial. To test this, we knocked down the expression of a subset of CIP-encoding genes using RNAi in the same C. elegans Aβ model and measured paralysis. In parallel, we performed the experiment with non-transgenic animals to rule out any non-specific effects on muscle function. We found that, of the 23 genes tested, 12 significantly impacted paralysis, eight had no impact, and three were either lethal or significantly delayed development and so could not be quantified (Fig. 4A). Therefore, of the CIP genes we were able to test, 60% significantly impacted Aβ toxicity (Fig. 4A Supp Tables 11A & B). Contrary to our initial hypothesis, however, most of these genes (7/12) exacerbated paralysis rather than preventing it. This may be because knocking down mRNA expression of some proteins could be further reducing the levels of the soluble protein thereby compounding loss of function due to insolubility. When compared against a knockdown screen of ~ 8000 protein-coding genes in a similar Aβ C. elegans model, our results represent a roughly 60-fold greater hit rate, suggesting that the CIP is highly enriched with disease-modifying proteins [55].
Fig. 4
Targeting the insoluble proteome genetically or pharmacologically modulates Aβ toxicity. A Statistically significant hits from RNAi paralysis screen against CIP proteins. Each point represents the relative proportion of paralyzed animals on that plate normalized to the average paralysis on the control vector for that experiment, approx. 40 animals per plate, three separate experiments, error bars = SEM, for statistical tests see Supplementary Table 11A. B Barplot of paralysis under 50 µM Urolithin A treatment, 40 animals per plate, three experiment replicates, error bars = SEM, unpaired t test. C Boxplot of tetramethylrhodamine, methyl ester (TMRM), mitochondrial membrane potential stain in Aβ expressing animals normalized to body area after pre-treatment with 50 μM Urolithin A, 40–50 animals per condition, bars = min. to max., unpaired t test. D Barplot of survival of Aβ expressing animals in 50 µM rotenone solution after pre-treatment with 50 µM Urolithin A, approx. 75 animals per condition, error bars = SEM, two-way ANOVA with Šídák’s multiple comparisons test
Since we observed strong evidence of mitochondrial protein unfolding in the insoluble proteome, we hypothesized that the CIP could be targeted by pharmacologically enhancing mitophagy with a small molecule. Urolithin A (UA) is a mitophagy-inducing, natural product derived from the bacterial metabolism of foods containing ellagitannins, such as pomegranate seeds and walnuts, in the gastrointestinal tract [56,57,58]. UA increases lifespan and protects neurons from amyloid toxicity in C. elegans through a mitophagy-dependent mechanism [59, 60]. UA supplementation in older adult humans was shown to improve muscle function in a phase II clinical trial for age-related frailty [61, 62]. We treated Aβ-expressing animals with UA for 18 h prior to inducing Aβ expression and scored paralysis after 24 h. We measured a robust and significant decrease in paralysis (Fig. 4B, Supp. Table 12). To confirm that UA improved mitochondrial quality we assayed mitochondrial function in Aβ expressing animals. Previous work demonstrated that Aβ expression caused a reduction in mitochondrial membrane potential [63], which we found to be significantly rescued by UA treatment (Fig. 4C). Additionally, we found that UA increased resistance to the mitochondrial complex I-specific inhibitor, rotenone (Fig. 4D). Taken with recent Aβ mouse model data [64], these results support that targeting mitochondrial quality control by mitophagy represents a reasonable strategy for preventing or treating Aβ toxicity.
留言 (0)