
記住我
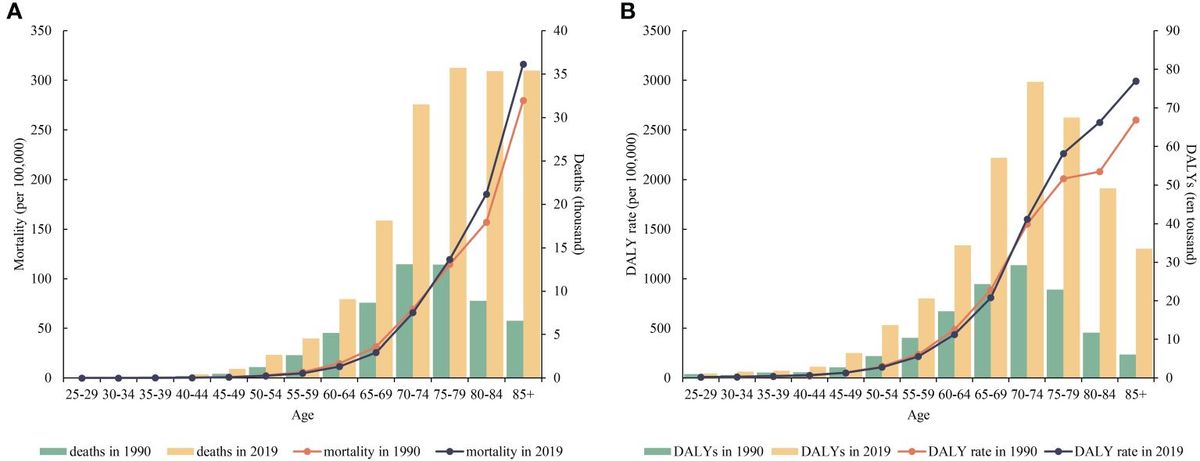

PCOS is a complex endocrine and metabolic disorder (1). The primary characteristics of PCOS patients are long-term anovulation or infrequent ovulation, insulin resistance (IR), hyperandrogenemia, polycystic ovarian morphology, and decreased female fertility (2–4). In addition to having abnormal reproductive function, many PCOS patients suffer from metabolic syndrome, IR, impaired glucose tolerance, obesity, atherosclerosis, and other metabolic abnormalities (5). At present, it is believed that the occurrence of PCOS is mainly related to genetic factors, environmental factors, and metabolic factors (6, 7). The specific causes of PCOS have not been fully elucidated (8, 9). Ferroptosis is caused by iron buildup and lipid peroxidation. It differs from cell apoptosis and necrosis (10). Ferroptosis is involved in regulating the occurrence and progression of diseases such as cancer, respiratory system diseases, cardiovascular system diseases, diabetes mellitus, and urinary system diseases and plays a key role in disease treatment (11) (Figure 1). There is limited and incomplete research on ferroptosis in the reproductive system. The role of ferroptosis in the reproductive system deserves further investigation (12, 13). Ferroptosis in PCOS is accompanied by the dysregulation of mitochondrial dynamics, the promotion of an inflammatory response, and the intensification of oxidative stress (14–16). This article reviews the correlation between ferroptosis and PCOS, providing ideas for exploring the underlying mechanisms of PCOS.
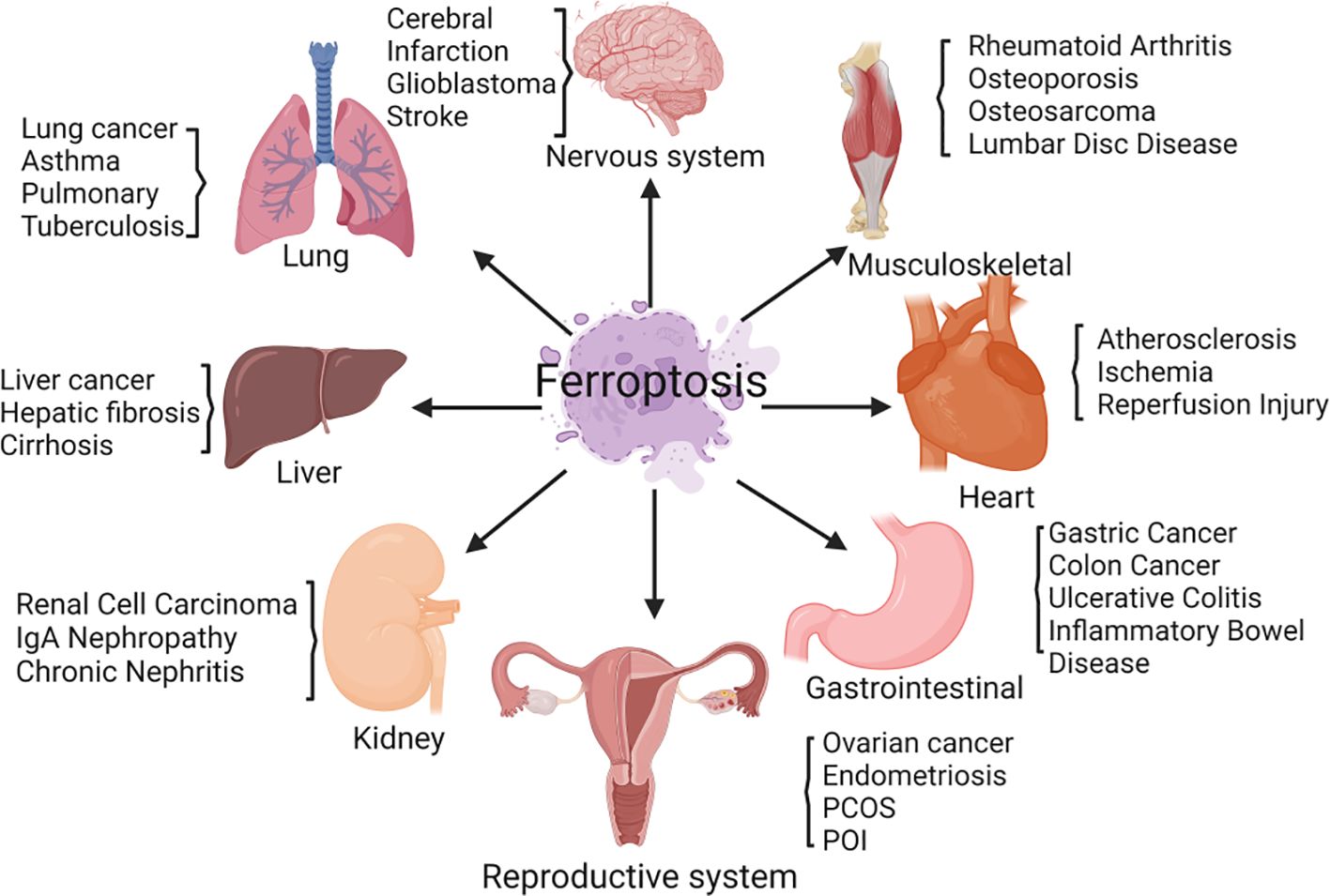
Figure 1 Ferroptosis is involved in regulating the occurrence and progression of various diseases, including those of the nervous system, respiratory system, cardiovascular system, diabetes mellitus, urinary system, and female reproductive system.
2 FerroptosisIn 2012, Dixon et al. proposed ferroptosis as a novel iron-dependent programmed cell death mechanism that is distinct from apoptosis, necrosis, and autophagy (17) (Table 1). Its main characteristics are as follows (1): During the process of cell death, a large amount of iron accumulates, and lipid peroxidation occurs, activating signaling pathways such as oxidative stress (18) (2); In the ultrastructure, cell atrophy, membrane rupture, and mitochondrial membrane wrinkling, and no significant nuclear morphological changes are observed (19). Ferroptosis is a nonenzymatic and enzymatic reaction that occurs under iron catalysis, leading to lipid peroxidation of cell membranes (20). Polyunsaturated fatty acids (PUFAs) are the main targets of lipid peroxidation of cell membranes (21). Glutathione peroxidase 4 (GPX4) regulates ferroptosis (22). Under antioxidant conditions, GPX4 can reduce the accumulation of intracellular reactive oxygen species (ROS) and reduce the sensitivity of cells to ferroptosis, thus affecting the occurrence of ferroptosis (23).
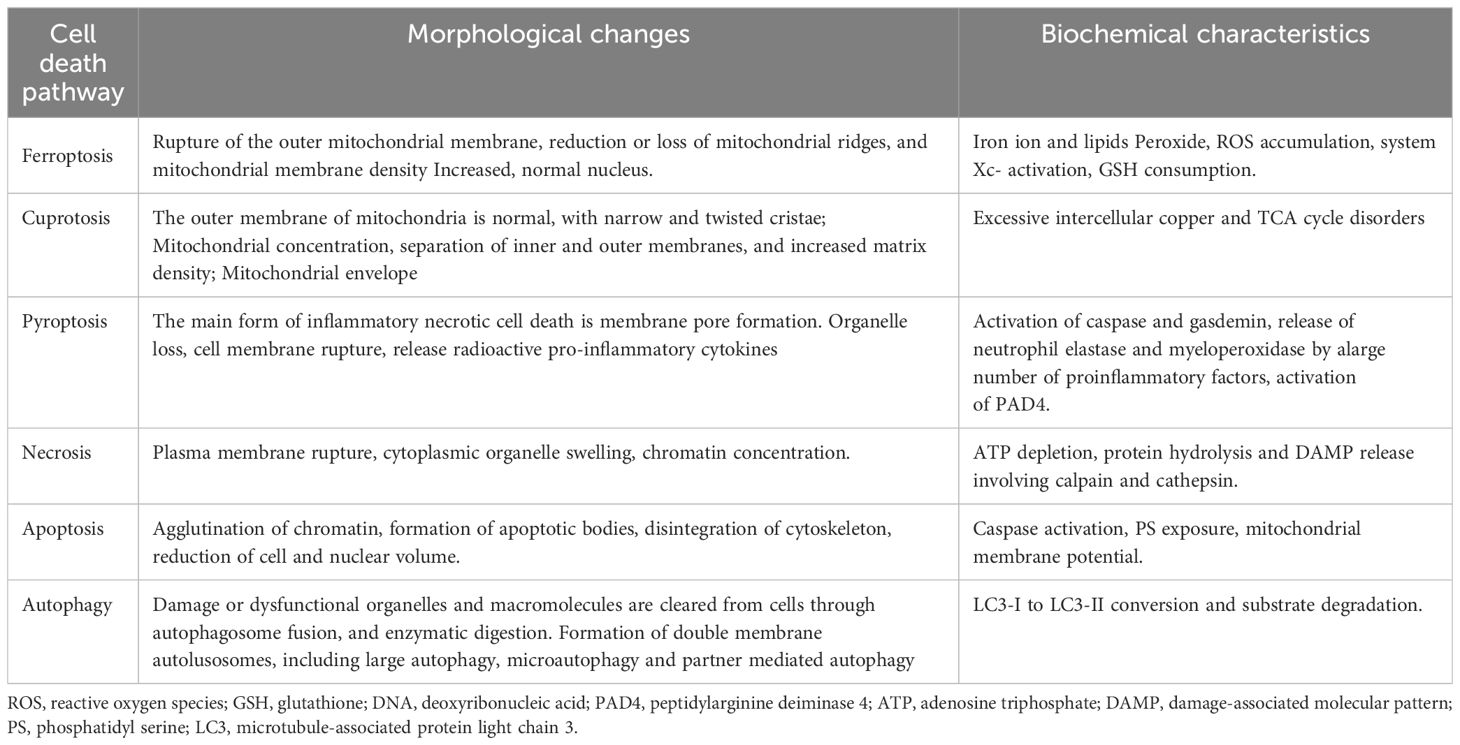
Table 1 Comparison of cell death pathways.
The Fenton reaction first removes a hydrogen atom from poly-unsaturated fatty acid-phosphatidyl ethanolamine (24). This leads to the formation of carbon-centered phospholipids, which then react with molecular oxygen to generate PLOO·, which can remove hydrogen from another PUFA and form phospholipid hydroperoxides (25). If antioxidants such as GPX4 are unable to promptly convert phospholipid hydroperoxides to the appropriate alcohols, phospholipid hydroperoxides and lipid free radicals will react with polyunsaturated fatty acid-phosphatidyl ethanolamine to further promote the production of phospholipid hydroperoxides (26). Ultimately, this leads to the production of many secondary products, such as lipid peroxidation products, leading to ferroptosis (27). Ferroptosis mainly causes oxidative metabolic disorders of cell membrane phospholipids through iron metabolism, lipid metabolism, and amino acid metabolism (28) (Figure 2).
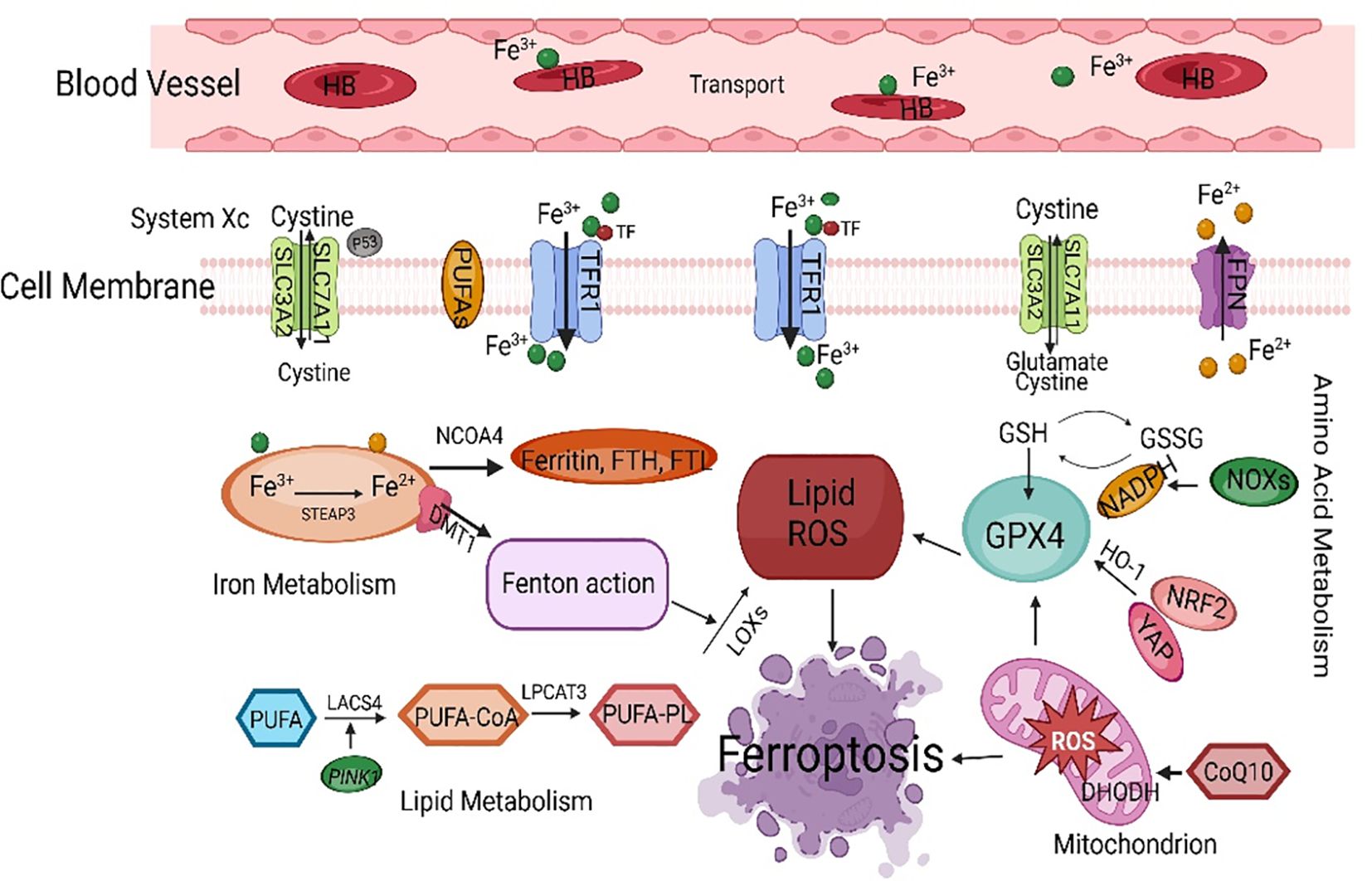
Figure 2 The main regulatory mechanisms of ferroptosis. SLC7A11, solute carrier family 7 member 11; PUFAs, polyunsaturated fatty acids; TFCR1, transferrin receptor 1; FPN, ferroportin; NCOA4, nuclear receptor coactivator 4; FTH, ferritin heavy chain; FTL, ferritin light chain; GSH, glutathione; GPX4, glutathione peroxidase 4; HO-1, heme oxygenase-1; NRF2, nuclear factor-erythroid 2-related factor 2; ROS, reactive oxygen species; LACS4, acyl-CoA synthetase long-chain family member 4.
2.1 Ferroptosis and iron metabolism2.1.1 Iron uptake, storage, and releaseFerroptosis requires the involvement of iron (29). The sensitivity of cells to ferroptosis depends on the input, output, storage, and transport of iron. There are two forms of iron in cells: Fe2+ and Fe3+ (30). A large amount of ROS is generated during the mutual conversion of Fe3+ and Fe2+ through redox reactions, leading to tissue damage and ferroptosis (31). Fe3+ has strong stability and is responsible for the storage and transportation of iron in the body; Fe2+ has an electron transport ability, and proteins containing Fe2+ can participate in various redox reactions (32). Transferrin (TF) carries Fe3+ in the bloodstream and transfers iron in cells through transferrin receptor 1 (TFCR1). Fe3+ is reduced to Fe2+ by the six-transmembrane epithelial antigen of prostate 3, which is stored in the ferritin heavy chain (FTH) and ferritin light chain (FTL) (33). Fe2+ is stored in ferritin (Fn) and becomes a part of the labile iron pool, which plays a dominant role in ferroptosis. TFCR1 is considered a marker protein for ferroptosis, and knocking out TFCR1 can block ferroptosis. In the absence of enough iron in cells, an “iron starvation response” begins, increasing iron availability by increasing TFCR1 and decreasing the levels of FTH and ferroportin (FPN) (34).
2.1.2 Iron homeostasisUnder normal circumstances, intracellular iron maintains a subtle balance through the iron transport system (35). Some iron is used for the biosynthesis of hemoglobin and iron sulfur clusters and for DNA synthesis. Cellular iron homeostasis also depends on iron regulatory proteins and iron-responsive elements (36). Iron regulatory proteins can bind to iron-responsive element mRNAs, regulating their translation process. Aberrant expression or dysfunction of the divalent metal transporter 1, TFRC1, and Fn genes and FPN leads to intracellular iron imbalance (37). Iron regulatory protein 2 can enhance the sensitivity of cells to erastin-induced ferroptosis by inhibiting the expression of FTH and FTL (38). When the plasma iron level meets the systemic iron demand, the liver will increase the secretion of hepcidin into the blood, reducing the plasma iron level and maintaining iron homeostasis in the body (39). Although every cell requires iron to generate energy, high iron levels can induce inflammation, oxidative stress, and lipid peroxidation in the cell membrane, leading to ferroptosis (40).
Iron homeostasis dysregulation is involved in ferroptosis, and multiple iron metabolism regulatory factors work together to maintain iron homeostasis (41). As mentioned, Fn autophagy is crucial for regulating ferroptosis (42). Nuclear receptor coactivator 4 (NCOA4) helps lysosomes breakdown intracellular Fn through autophagy, which releases free iron and causes oxidative damage (43). This selective autophagy process is called iron autophagy. Knockout of NCOA4 can inhibit ferroptosis caused by decreased amino acid or cysteine levels (44). Research has shown that knocking out autophagy-related genes 5 (ATG5) and ATG7 can reduce intracellular Fe2+ levels and lipid peroxidation, inhibiting ferroptosis (45). In addition to selective autophagy, activating the ubiquitin−proteasome system can promote Fn degradation. As an antioxidant, ubiquitin captures lipid peroxidation free radicals, prevents lipid peroxidation generation, and inhibits ferroptosis. Tang et al. reported that in treated rat hearts, ubiquitin-specific protease 7 increases iron uptake and promotes ferroptosis by activating the P53/TFRC1 pathway (46).
2.2 Ferroptosis and lipid metabolismThe conversion of lipids into membrane phospholipids and the occurrence of peroxidation are necessary conditions for ferroptosis (47). Lipid metabolism is closely related to ferroptosis. Most ROS related to ferroptosis originates from the Fenton and Haber-Weiss reactions, which then interact with PUFAs on the lipid membrane to form ROS (48). Phosphatidylethanolamine and arachidonic acid are essential membrane phospholipids that cause ferroptosis in the cell membrane. Lipoxygenases are nonheme iron-dependent dioxygenases that target polyunsaturated fatty acids (PUFAs). With the participation of iron in the cytoplasm, lipid free radicals are formed, which can directly oxidize PUFAs and PUFA-containing lipids in the biofilm, leading to cell damage (49). A decrease in lipoxygenase expression can also effectively improve ferroptosis induced by erastin (50). However, lipoxygenases cannot prevent ferroptosis induced by RSL3 (51). Although lipoxygenases do not have extensive regulatory effects on ferroptosis, they play essential roles in specific ferroptosis mechanisms.
Acyl-CoA synthetase long chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 are key enzymes involved in the process of lipid peroxidation (52). ACSL4 and lysophosphatidylcholine acyltransferase 3, which are involved in the synthesis of PUFAs, play essential roles in the ferroptosis pathway (53). ACSL4 binds PUFA to coenzyme A through acylation and further undergoes an esterification reaction with phosphatidylethanolamine under the action of LPCAT to generate PUFA-PE (54). Continuous oxidation reactions and consumption of PUFAs may alter the fluid structure of cell membranes, thereby increasing membrane permeability and ultimately leading to cell death. Inhibiting the expression of ACSL4 can increase the resistance of cells to ferroptosis and can be a target for inhibiting ferroptosis, which may provide new ideas for diagnosing and treating PCOS. Ferroptosis may play a role not only in pathological conditions but also in physiological processes. The normal development of the human fetal immune system depends on sufficient dietary intake of PUFAs (55). To summarize, studying the correlation between lipid metabolism and ferroptosis has essential research value (56).
2.3 Ferroptosis and amino acid metabolismFerroptosis is closely related to amino acid metabolism (57). The consumption of glutathione (GSH) can lead to the inactivation of GPX4 (58). Cysteine synthesizes GSH in the body through the cystine/glutamic acid reverse transporter (System Xc -) and the transsulfuration pathway (59). System Xc -, located on the cell membrane, is a heterodimer linked by disulfide bonds and contains the heavy chain subunits solute carrier family 3 member 2 and solute carrier family 7 member 11 (SLC7A11) (60). System Xc - is responsible for transporting extracellular cysteine into the cell. Glutamate exchanges cysteine at a 1:1 ratio, transporting intracellular glutamate to the outside of the cell (61). Cysteine provides the raw material for intracellular GSH synthesis (62). Another source of cysteine is cysteine sulfide, which is formed through the reverse sulfurization of methionine (63).
2.3.1 Negative regulatory factors of System Xc -Inhibiting cysteine uptake by System Xc - can inhibit the synthesis of GSH, leading to the accumulation of peroxides in the body and the induction of ferroptosis (64). System Xc—inhibitors such as erastin, sulfasalazine, sorafenib, and glutamate—are considered class I ferroptosis inducers, and inhibition of System Xc - leads to compensatory transcriptional upregulation of SLC7A11 (65). SLC7A11, an essential component of System Xc -, is an upstream regulatory factor of ferroptosis (66). SLC7A11 can reduce lipid peroxidation accumulation and prevent cells from entering the ferroptosis program by introducing cysteine and promoting GSH synthesis (67). Song et al. reported that Beclin 1 directly stops System Xc - activity by attaching to the core region of SLC7A11 and encouraging ferroptosis (68). Nuclear factor erythroid 2-related factor 2 (NRF2) is a crucial antioxidant transcription factor. Silencing NRF2 can significantly reduce the expression of SLC7A11 and heme oxygenase-1 (HO-1) and inhibit ferroptosis (69).
2.3.2 GPX4: a core regulator of ferroptosisFerroptosis is related to the inactivation of GPX4 (70). As an important antioxidant in cells, GPX4 is a key ferroptosis regulator. GSH is used as a reducing substrate to promote the transformation of lipid peroxidation products into hydroxyl compounds, preventing ferroptosis in cells and protecting the structure and function of cell membranes from interference and damage (71). Under normal circumstances, fatty acid hydroperoxides can be converted into fatty acid alcohols through GPX4 mediation (72). Inhibiting GPX4 can interfere with intracellular iron homeostasis and reduce lipid peroxidation. The ferroptosis inducers RSL3 and erastin can both inactivate GPX4. RSL-3 blocks GPX4, which increases the levels of ROS and malondialdehyde (MDA) inside cells and promotes ferroptosis by blocking the SLC7A11/GSH/GPX4 pathway (73). Erastin indirectly inhibits GPX4 by inhibiting cysteine input, leading to cell membrane damage and death. Selenocysteine is an amino acid found in the active center of GPX4 (74). It can maintain GPX4 activity and help eliminate lipid peroxidation, which stops ferroptosis. Therefore, selenium deficiency can inhibit GPX4 activity and induce ferroptosis. GPX4 can also be turned off directly by squalene synthase, HMG CoA reductase, and other enzymes, which can change reduction reactions (75).
2.4 Ferroptosis and antioxidant pathwaysUnder physiological conditions, when cells undergo lipid peroxidation, multiple antioxidant pathways counteract this change. Research has shown that the intracellular ferroptosis antioxidant system is related to the SLC7A11/GSH/GPX4 signaling pathway (76). GPX4 reacts with GSH and lipid peroxidation products, efficiently clearing accumulated lipid peroxidation products and maintaining normal physiological functions. Ferroptosis suppressor protein 1 (FSP1) is a newly discovered inhibitory protein that inhibits GPX4 deficiency (77). The NAD(P)H/CoQ10/FSP1 signaling pathway is a crucial ferroptosis regulatory pathway (78). GTP cyclohydrolase 1 is involved in ferroptosis resistance, and its mechanism involves the generation of tetrahydrobipterin (BH4), which has redox activity, from GTP (79). BH4, a powerful antioxidant that captures free radicals, can promote CoQH2 regeneration and combat lipid peroxidation by activating downstream molecules such as GTP cyclohydrolase 1. These results indicate that the NADH-FSP1 CoQ10 and GTP cyclohydrolase 1/BH4 pathways work together with the SLC7A11/GPX4/GSH pathway to inhibit ferroptosis (80). Mao et al. reported that there is a ferroptosis system in mitochondria mainly composed of dihydrolactate dehydrogenase, which also induces lipid peroxidation in the mitochondrial membrane by promoting the reduction of CoQ10 to combat ferroptosis (81).
2.5 Ferroptosis and other regulatory mechanisms2.5.1 p53-mediated ferroptosisResearch has shown that p53 has a complex dual mechanism for regulating cellular ferroptosis through transcription and translation (82). P53 can inhibit cysteine uptake by downregulating SLC7A11, reducing GPX activity and GSH synthesis and subsequently causing intracellular ROS accumulation and ferroptosis (83). On the other hand, p53 can promote the activity of glutaminase 2 and spermidine/spermine N1-acetyltransferase 1 (SAT1). Glutaminase 2 catalyzes the degradation of glutamine to glutamic acid, and the activation of SAT1 induces lipid peroxidation. The overexpression of glutaminase 2 and SAT1 promotes PUFA oxidation and lipid peroxidation, leading to ferroptosis. The transcription target gene cyclin-dependent kinase inhibitor 1A is used to reduce GSH and ROS and delay ferroptosis (84). The specific mechanism by which P53 regulates ferroptosis needs further study.
2.5.2 The Keap1-NRF2 pathway mediates ferroptosisThe NRF2 transcription factor stimulates the production of NADPH by blocking the expression of antioxidant genes and increasing the expression of enzymes in the pentose phosphate pathway, which decreases the sensitivity of cells to ferroptosis (85). Under normal physiological conditions, activation of the Keap1-NRF2 signaling pathway promotes the activation of System Xc - and the expression of GPX4 and accelerates cysteine glutamate transport, thereby clearing accumulated lipid peroxidation and inhibiting ferroptosis (86). Activation of NRF2 reduces iron absorption, limits ROS production, and enhances cellular antioxidant capacity (87). P62 strictly controls NRF2 and can inhibit ferroptosis. P62 is an autophagic receptor that directly inhibits Keap1 while promoting NRF2 activation (88). NRF2 regulates multiple targets, such as genes regulating GSH synthesis and encoding antioxidant proteins (89). The iron-chelating enzymes HO-1, FTH, and FTL are all strictly controlled by NRF2 (90). Glutamate cysteine ligase, GSH synthase, and SLC7A11 are also transcriptional targets of NRF2 (91).
2.6 The interaction between ferroptosis and other types of cell deathFerroptosis may interact with other types of cell death (92). Anthraquinone modifications can significantly upregulate the expression of glucose-regulated protein 78, activate transcription factor 4, and downregulate the expression of the essential ferroptosis protein GPX4. Endoplasmic reticulum stress induces apoptosis and accompanies ferroptosis. The autophagy pathway can degrade ferritin, while ferroptosis regulatory proteins can regulate autophagy (93). Ferritin autophagy and lipid autophagy can promote ferroptosis by regulating iron metabolism and lipid peroxidation (94). Under oxidative stress, autophagy protects mitochondrial integrity by clearing ROS, preventing cell apoptosis, and exerting a protective effect (95). Excessive ROS-induced autophagy can also lead to cell death (96). Lipid peroxidation can attach to particular mitochondria and autophagy-related proteins, leading to autophagic cell death and cellular dysfunction (97). In summary, ferroptosis interacts with and promotes other types of cell death.
3 The role of ferroptosis in female reproductive disorders3.1 The impact of ferroptosis on the process of follicular developmentROS and antioxidants in the ovaries play critical regulatory roles during oocyte maturation, fertilization, and embryonic development and implantation (98). Studies have shown that excessive iron in follicular fluid significantly increases ROS levels in mouse oocytes, leading to a decrease in the maturation rate of oocytes (99). Recurrent bleeding from ovarian lesions can lead to iron overload, increased ferroptosis, and decreased ovarian function, affecting follicular development and oocyte quality. ROS in follicles promotes apoptosis in most follicular cells (100). Studies have shown that blocking the production of GSH increases antral follicular atresia in rats (101). Research has shown that the expression of TF in early follicular atresia is significantly reduced, while the expression of the iron chaperone protein PCBP is increased considerably. During follicular development, Basonclin1 maintains lipid metabolism and redox homeostasis in oocytes. Research has confirmed abnormal levels and pathways of ferroptosis-related indicators in basonclin1-truncated mouse oocytes (102). Neurofibromin 2 expression was significantly reduced in basonclin1-deficient oocytes, while the expression levels of yes-associated protein, TFR, and ACSL4 increased considerably, triggering oocyte ferroptosis (103).
3.2 Ferroptosis of GCs leads to immature oocytesOogenesis is achieved through the interaction between the oocyte and the microenvironment of the follicle (104). The production of various cytokines and hormones in follicular fluid mainly relies on ovarian GCs, which play an essential role in oogenesis (105). Excessive iron in the internal environment can cause ferroptosis in ovarian GCs, which slows oocyte maturation and follicle development, increasing the risk of infertility (106, 107). Researchers have shown that a TFR-mediated increase in iron uptake in GCs induces the release of ROS, mitochondrial autophagy, and lipid peroxidation (108). The levels of FTH, TF, and TFRC in the ovarian GCs of infertile women are much lower than those in the ovarian GCs of healthy women (109). This finding suggested that FTH is a crucial regulator of ovarian follicle development and atresia (110). CircRHBG competes with SLC7A11 for binding to miR-515-5p, inhibiting GC ferroptosis. Therefore, the knockout of circRHBG can promote ferroptosis (111).
3.3 Oxidative stress impairs reproductive functionMitochondrial dysfunction and excessive ROS production are characteristics of ferroptosis, and mitochondrial dysfunction further promotes ROS production, leading to oxidative stress, which induces the development and exacerbation of ferroptosis (112, 113). The other key indicator of ferroptosis is an increase in the levels of MDA, the final product of lipid oxidation (114). MDA can affect the activity of respiratory chain complexes and critical enzymes in mitochondria and aggravate membrane damage. A reduction in mitochondrial DNA is correlated with IR, hyperandrogenemia, and polycystic ovarian morphology in women with PCOS (115–117). Oxidative stress, ferroptosis, and PCOS interact with each other (118). An imbalance in total antioxidant levels in the serum of women with PCOS can exacerbate cell damage and reduce cellular defense ability (119). By comparing the oxidative stress indices of women with PCOS and healthy women, it was found that women with PCOS have significantly greater GPX and GSH reductase activities (120). The concentration of these substances directly affects the maturation and quality of oocytes, fertilization, and embryonic development (121). Metabolomic analysis of follicular fluid from clinical PCOS patients revealed mitochondrial dysfunction, oxidation−reduction potential imbalance, and increased oxidative stress in cumulus cells (122). The elevated levels of autoantibodies and ROS in the serum of PCOS patients suggest that oxidative stress may be one of the critical reasons for the abnormal endometrial environment in PCOS patients (123). Insulin is the primary regulator of oxidative phosphorylation, and its secretion can directly affect mitochondrial function (124, 125).
3.4 The impact of ferroptosis on pregnancy outcomesIron homeostasis plays an essential role in maintaining pregnancy. In vitro experimental studies have shown that appropriate iron in protein-free embryo culture medium can promote embryonic development, while iron overload is not conducive to embryonic development. Ferroptosis is associated with placental injury or miscarriage (126). During the abortion process, the large amount of ROS generated increases the generation of lipid peroxidation at the maternal-fetal interface, providing a primary condition for ferroptosis. Ferroptosis is triggered in the uterus and placenta of PCOS rats with hyperandrogenemia and IR. Compared with those in the control group, the expression levels of GPX4 and GSH in the uterus and placenta of PCOS rats were lower, and the expression levels of the ferroptosis-related genes ACSL4, TFCR1, SLC7A11, and glutamate cysteine ligase C were significantly increased (127). Ferroptosis is closely related to spontaneous preterm birth, and PLA2G6 can alleviate ferroptosis caused by GPX4 inhibition during pregnancy in mice (128).
4 Ferroptosis in PCOSPCOS is a reproductive endocrine disorder that not only affects women’s reproductive and physiological health but also leads to complications such as diabetes mellitus type 2 (T2DM), obesity, familial cardiovascular disease, and cardiovascular disease (129). PCOS has transcended the field of obstetrics and gynecology and affects various significant systems throughout the body, seriously threatening women’s physical and mental health, affecting their quality of life, and causing lifelong endocrine and metabolic diseases (130, 131) (Figure 3). The etiology of PCOS has not yet been clarified, although much work has been done on this topic over the past few decades (132). The onset of PCOS involves multiple factors, such as hyperandrogenemia, IR, and abnormal follicular development (133–135). Ferroptosis affects the proliferation and apoptosis of granulosa cells (GCs), affecting endocrine and metabolic processes (Figure 4).
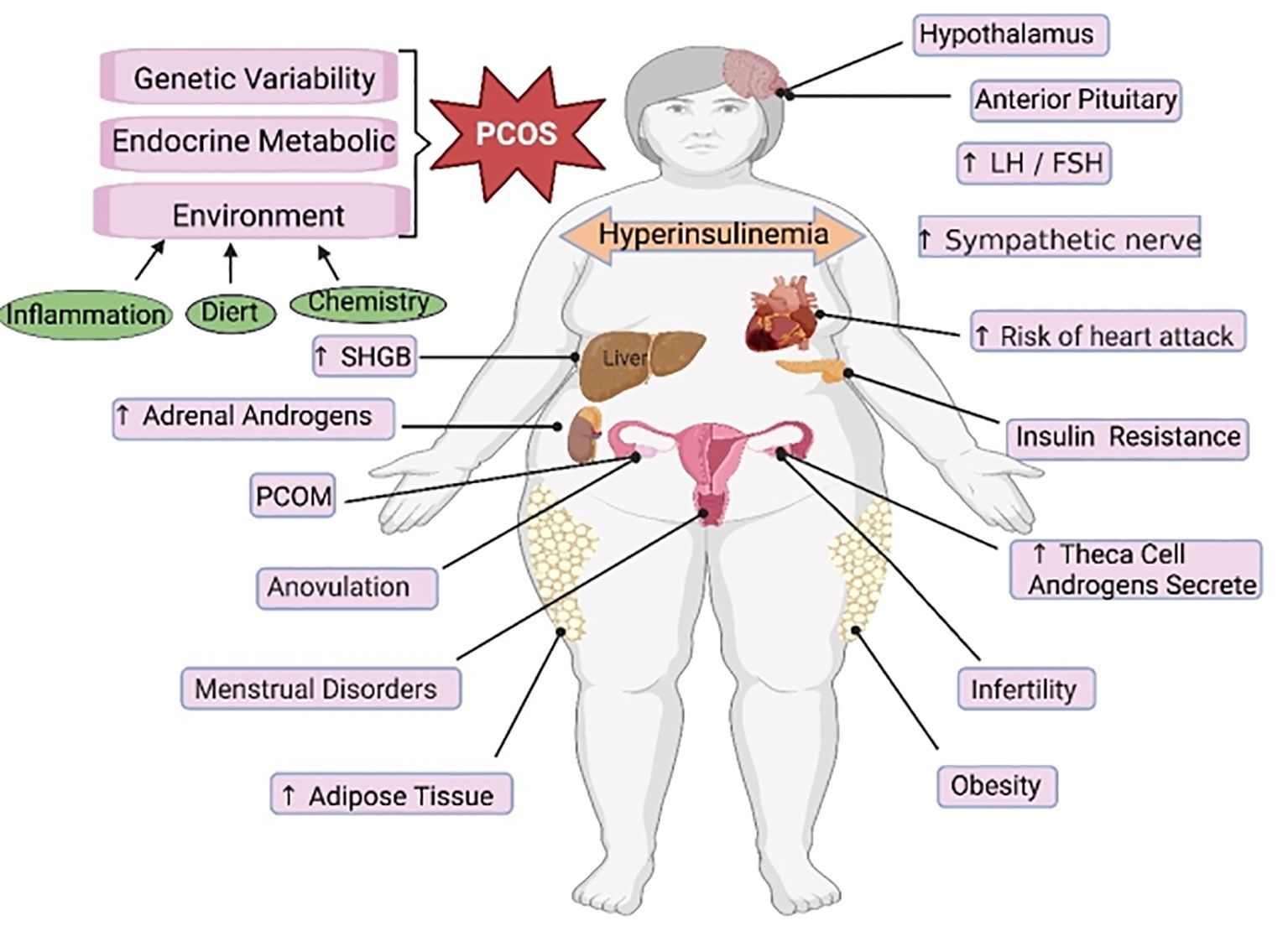
Figure 3 The main regulatory mechanisms of ferroptosis in PCOS. PCOS, polycystic ovarian syndrome; SHGB, sex hormone-binding globulin; LH, luteinizing hormone; FSH, follicle-stimulating hormone.
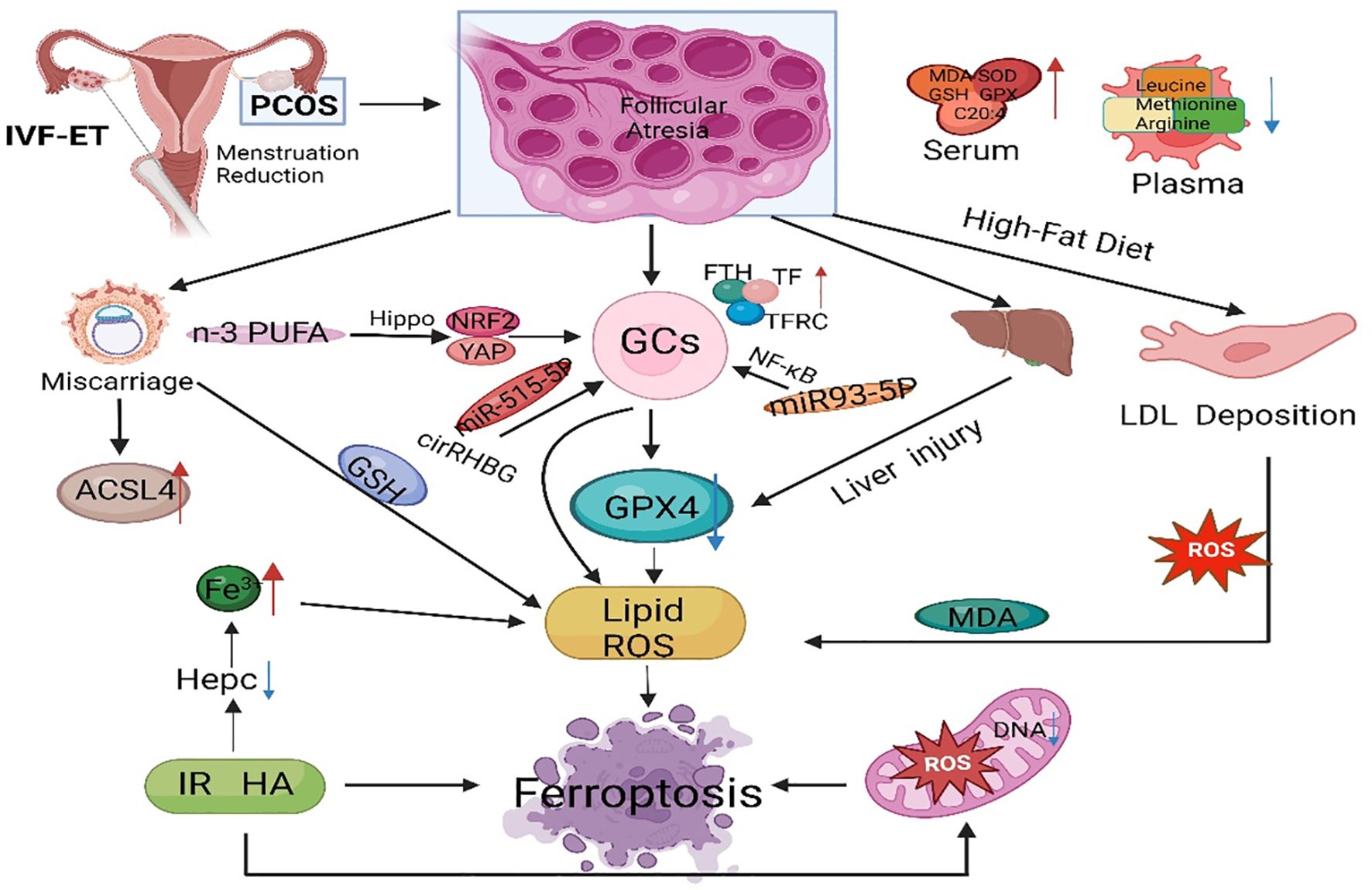
Figure 4 The main regulatory mechanisms of ferroptosis in PCOS. PCOS, polycystic ovarian syndrome; GCs, granulosa cells; GPX4, glutathione peroxidase 4; FTH, ferritin heavy chain; TF, transferrin; TFRC, transferrin receptor; IVF-ET, in vitro fertilization and embryo transfer; PUFA, polyunsaturated fatty acid; ACSL4, acyl-CoA synthetase long chain family member 4; IR, insulin resistance; ROS, reactive oxygen species; MDA, malondialdehyde; GSH, glutathione; NRF2, nuclear factor-erythroid 2-related factor 2.
Iron is an essential trace element for life and an important component of hemoglobin, myoglobin, and various enzymes (136). Iron participates in multiple critical physiological and biochemical processes in the body, including oxygen transport, DNA biosynthesis, and ATP synthesis (137). Research has shown that regardless of obesity, the serum Fn levels in PCOS patients are significantly greater than those in control individuals, indicating that abnormal iron metabolism may be involved in the occurrence and development of PCOS (138). Iron overload occurs in PCOS patients, potentially due to chronic oligomenorrhea and decreased hepcidin concentration (139). Research has shown that the serum Fn concentration is directly proportional to the severity of menstrual dysfunction, suggesting that the iron retention effect of chronic oligomenorrhea may be associated with increased iron storage in some PCOS patients (140). The compensatory hyperinsulinemia caused by PCOS may promote iron absorption (141, 142). Additionally, with the combined action of IR and excessive androgen in PCOS patients, TF inhibition is decreased, iron absorption in the intestine is increased, and iron release from macrophages is decreased, leading to iron overload (143).
4.1 Ferroptosis is involved in endocrine and metabolic disorders in PCOSIron overload can affect glucose metabolism, leading to IR and exacerbating metabolic abnormalities in PCOS patients (144). On the one hand, iron can affect the secretion and sensitivity of insulin and inhibit liver glycogen production, and insulin secretion decreases with increasing liver iron storage, leading to systemic hyperinsulinemia. On the other hand, insulin can promote iron absorption and Fn synthesis while also promoting glucose transport. Excessive androgen and insulin resistance can activate ferroptosis in the uterus and placenta of pregnant women with PCOS (145). The plasma levels of leucine, isoleucine, methionine, glutamine, and arginine were much lower in PCOS patients than in healthy controls (146). Compared to those in healthy women, serum bioactive lipid levels are much lower in women with PCOS (147). Arachidonic acid levels in the serum of PCOS rats were significantly greater than those in the serum of control rats (148). Phosphatidylethanolamine combined with arachidonic acid is an essential phospholipid that causes ferroptosis, which means that there are appropriate conditions for ferroptosis in PCOS patients (149).
4.2 Oxidative stress mediates metabolic abnormalities in PCOSOxidative stress is closely related to obesity, IR, and HA in PCOS, and the induced apoptosis of ovarian GCs leads to follicular atresia, which is one of the mechanisms of ovulation disorders in PCOS (150). SOD is an important antioxidant enzyme for scavenging oxygen-free radicals and catalyzes the dismutation reaction of ROS to eliminate oxygen-free radicals and reduce damage to ovarian cells (151). MDA levels are positively correlated with BMI, triglyceride levels, low-density lipoprotein levels, systolic blood pressure, diastolic blood pressure, and HOMA-IR (152). Research has shown that ROS produced by monocytes in the pancreas plays an essential role in the abnormal development of cells (153). ROS-induced NF-κB can enter the nucleus and bind to chromatin to promote tumor necrosis factor-α (TNF-α) (154). Transcription can activate the PI3K/Akt/mTOR pathway to inhibit insulin secretion (155). In PCOS animal models, free fatty acids in ovarian tissue increase, glucose oxidation is inhibited, free fatty acid oxidation is enhanced, and ROS increase through the TCA cycle, leading to IR (156).
4.3 Ferroptosis and a high-fat dietObesity is closely related to PCOS and plays a crucial role in the occurrence and development of PCOS (157). In obese PCOS patients, long-term high-fat diets can induce inflammatory reactions and oxidative damage and exacerbate iron deposition, which is an essential factor influencing ferroptosis (158). A long-term high-fat diet can cause low-density lipoprotein, which is deposited in the endothelium of tissues. The prolonged accumulation of low-density lipoprotein can induce an oxidative stress response (159). Oxidative stress is one of the pathogenic mechanisms of PCOS, and many studies have confirmed that oxidative stress indicators are positively correlated with obesity. Compared with those in nonobese patients, the serum levels of the antioxidant substances MDA, SOD, GSH, and GPX were significantly greater in the obese PCOS group (160). In the environment of mature follicles, oxidative damage can further trigger chronic inflammatory reactions in the ovaries. ROS plays a crucial role in follicular development disorders (161). In follicular fluid containing high levels of ROS, a large amount of highly toxic MDA is produced, which can exacerbate cell damage in the ovaries. Therefore, obesity, ferroptosis, and the development of PCOS are closely related.
5 Ferroptosis contributes to the diagnosis and treatment of PCOSFollicular development is a complex biological process of continuous change involving multiple hormones and regulatory factors, and the loss of oocytes from the ovary is irreversible (162). Abnormal follicular development can cause a wide range of female disorders and lead to reduced female fertility (163). Early preventive measures could mitigate the development of PCOS (164). The substances that regulate ferroptosis can serve as targets for diagnosing and treating PCOS, exhibiting broad research prospects. Zheng et al. reported that unexplained liver injury in PCOS patients and animal models was accompanied by increased Fe deposition and downregulation of hepcidin and GPX4 expression in the liver, indicating the importance of iron metabolism in this type of unexplained liver injury (165). Tang et al. reported that NEDD4L promotes ferroptosis in GCs and promotes the occurrence of PCOS by promoting GPX4 ubiquitination and degradation. NEDD4L decreased the viability of KGN cells and increased the levels of MDA and ROS. Moreover, ferroptosis inhibitors can block NEDD4L-induced KGN cell death, suggesting that NEDD4L regulates ferroptosis in KGN cells (166). Jiang et al. reported that KGN cells treated with DHEA exhibited ferroptosis characterized by decreased viability, inhibited GPX4 and SLC7A11 expression, increased ACSL4 expression, increased MDA levels and ROS accumulation, and increased lipid peroxidation. These findings may provide new insights into the pathophysiology and treatment of PCOS (167).
Zhang et al. noted that differentially expressed ferroptosis-related genes are associated with reproductive outcomes in infertile POCS patients, and they constructed a FerSig risk prognosis model (168). Lin et al. identified five essential differentially expressed ferroptosis-related genes (NOX1, ACVR1B, PHF21A, FTL, and GALNT14) that may be related to the pathogenesis of PCOS, providing a new perspective for the clinical diagnosis and treatment of PCOS (169). Research has shown that miR-93-5p regulates the NF-κB signaling pathway and promotes apoptosis and ferroptosis in GCs (170). Silencing miR-93-5p can prevent GC dysfunction and provide new molecular targets for diagnosing and treating PCOS. N-3 PUFAs activate Hippo, promote yes-associated protein 1 exocytosis, weaken cross-talk between yes-associated protein 1 and Nrf2, and ultimately activate ferroptosis sensitivity in ovarian GCs. N-3 PUFAs also inhibit excessive proliferation of GCs in ovarian follicles, by which n-3 PUFAs weaken PCOS, and identifying yes-associated protein 1 (Nrf2) as a potential therapeutic target for regulating GCs in PCOS (171). Transferrin receptor-mediated ROS promote ferroptosis in KGN cells by regulating NADPH oxidase 1/PTEN-induced kinase 1/acyl-CoA synthetase long-chain family member 4 signaling, and the inhibitory effects of TFRC/NOX1/PINK1/ACSL4 signaling on folliculogenesis could be a potential target for PCOS treatment (172). Peng et al. reported that metformin regulates ferroptosis through the SIRT3/AMPK/mTOR pathway to improve weight, metabolic disorders, and ovarian dysfunction in PCOS mice (173). Therefore, exploring the role of ferroptosis in the occurrence and development of PCOS can provide more ideas for mechanistic research on PCOS and more potential targets for PCOS treatment.
6 ConclusionsIn recent years, substantial progress has been made in exploring ferroptosis. The regulatory network of iron homeostasis, lipid metabolism, amino acid metabolism, and antioxidant pathways provides new ideas for diagnosing and treating human diseases. Ferroptosis is expected to become a new biomarker for the development, treatment efficacy, and prognostic evaluation of PCOS. Research on ferroptosis in PCOS is still lacking. Further research on PCOS animal models with larger sample sizes is needed to validate the potential effects of ferroptosis on ovarian GCs, follicles, ovaries, and even the entire female reproductive system. More clinical studies are urgently needed. Assessing whether ferroptosis and its related molecules play essential roles in infertility, metabolic abnormalities, and other aspects of clinical PCOS and whether the administration of antioxidants can prevent ferroptosis, lipid peroxidation, and adverse maternal and fetal outcomes caused by maternal hyperandrogenemia and IR will provide insights and directions for future clinical diagnosis and treatment. Based on existing prevention and treatment methods for PCOS, interventions targeting different nodes of the ferroptosis regulatory network combined with other treatment methods for various patient etiologies, treatments, and prognoses are expected to be effective and reasonable in the future, thus achieving personalized treatment of PCOS.
Author contributionsMW: Conceptualization, Formal analysis, Methodology, Project administration, Resources, Writing – original draft, Writing – review & editing. B-QZ: Conceptualization, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. SM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Resources, Writing – review & editing. YX: Formal analysis, Funding acquisition, Investigation, Methodology, Software, Writing – review & editing. D-HZ: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Writing – original draft. J-SZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Software, Writing – review & editing. C-JL: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Resources, Visualization, Writing – review & editing. XZ: Conceptualization, Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. L-WZ: Conceptualization, Investigation, Supervision, Validation, Visualization, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Natural Science Foundation of Jilin Province (NO YDZJ202301ZYTS434, NO YDZJ202201ZYTS007) and the Jilin Provincial Development and Reform Commission Project (2023C037-4).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
1. Bell RJ, Islam RM, Skiba MA, Davis SR. Reply: A single cut-off value of anti-Mullerian hormone should not be used for the diagnosis of PCOS in all reproductive-aged women. Hum Reprod. (2022) 37:622. doi: 10.1093/humrep/deac013
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Cooney LG, Lee I, Sammel MD, Dokras A. High prevalence of moderate and severe depressive and anxiety symptoms in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod. (2017) 32:1075–91. doi: 10.1093/humrep/dex044
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Walters KA, Bertoldo MJ, Handelsman DJ. Evidence from animal models on the pathogenesis of PCOS. Best Pract Res Clin Endocrinol Metab. (2018) 32:271–81. doi: 10.1016/j.beem.2018.03.008
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Tay CT, Teede HJ, Hill B, Loxton D, Joham AE. Increased prevalence of eating disorders, low self-esteem, and psychological distress in women with polycystic ovary syndrome: a community-based cohort study. Fertil Steril. (2019) 112:353–61. doi: 10.1016/j.fertnstert.2019.03.027
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. (2014) 16:1180–91. doi: 10.1038/ncb3064
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Banuls C, Rovira-Llopis S, Martinez de Maranon A, Veses S, Jover A, Gomez M, et al. Metabolic syndrome enhances endoplasmic reticulum, oxidative stress, and leukocyte-endothelium interactions in PCOS. Metabolism. (2017) 71:153–62. doi: 10.1016/j.metabol.2017.02.012
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Lai Q, Xiang W, Li Q, Zhang H, Li Y, Zhu G, et al. Oxidative stress in granulosa cells contributes to poor oocyte quality and IVF-ET outcomes in women with polycystic ovary syndrome. Front Med. (2018) 12:518–24. doi: 10.1007/s11684-017-0575-y
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. (2021) 81:355–69 e10. doi: 10.1016/j.molcel.2020.11.024
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Nasri F, Zare M, Doroudchi M, Gharesi-Fard B. Proteome analysis of CD4(+) T cells reveals differentially expressed proteins in infertile polycystic ovary syndrome patients. Endocr Metab Immune Disord Drug Targets. (2021) 21:1998–2004. doi: 10.2174/1871530320666201119152323
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Wen J, Chen H, Ren Z, Zhang P, Chen J, Jiang S. Ultrasmall iron oxide nanoparticles induced ferroptosis via Beclin1/ATG5-dependent autophagy pathway. Nano Converg. (2021) 8:10. doi: 10.1186/s40580-021-00260-z
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of poly-unsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
CrossRef Full Text | Google Scholar
22. Li C, Deng X, Zhang W, Xie X, Conrad M, Liu Y, et al. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J Med Chem. (2019) 62:266–75. doi: 10.1021/acs.jmedchem.8b00315
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. (2020) 585:603–8. doi: 10.1038/s41586-020-2732-8
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. (2020) 16:302–9. doi: 10.1038/s41589-020-0472-6
留言 (0)