
記住我





The adapted PCR protocol allowed us to screen all DNA samples of family members and individual patients. Repeat length differences of control samples and patients could be seen on agarose control gels, already (Fig. 2a). Capillary gel electrophoresis of PCR products allowed us to determine the correct repeat sizes for all tested patients and controls (Fig. 2b and c).
The largest expansion in our cohort is composed by 68 GGC triplets in patient 8 (Table 2) with an AOO of 16 years, the shortest pathological repeat in our collective turned out to be 44 repeats with an AOO of about 60 years, respectively (Table 2; P9).
Sanger sequencing of control and patient samples could confirm the repeat lengths and structures. Common normal alleles are composed of 21 triplet units, whereas heterozygous controls with alleles of 18/21 or 19/21 repeats are also present. The normal 21 allele typically has a structure build of 6 and 14 glycine coding elements interrupted by an AGT serine coding triplet at position 7 (Fig. 2d). At position 5 and 13, the GGC stretches are interrupted by synonymous glycine coding GGT triplets. Length differences are due to the loss of two or three GGC units, respectively. Interestingly, the 19 repeat allele (allele 2) misses the synonymous GGT interruption at position 13.
The same sequencing approach was applied to the mixture of normal and expanded alleles in affected patients. By extracting separated PCR bands from the agarose gel, we were able to generate clear signals for the smaller normal allele and heterozygous signals for expanded alleles at the nucleotide positions, where the above-mentioned interruptions are located (Online Resource 1, Fig. 1). These results underline the fact that all interruptions are lost in expanded alleles, indicating their potential role as stabilizing elements in normal alleles.
Within the two families, we observed (i) strong anticipation (Fig. 3) from generation to generation with relatively small differences in repeat lengths, and (ii) a negative correlation between the repeat length of the longer allele and AOO (Pearson correlation coefficient R = − 0.901, R2 = 0.812, p < 0.001) explaining of the variability of AOO. The data were normally distributed (Shapiro Wilk test statistic 0.981, p = 0.889). In family I we observed a decrease of AOO from 50 to 21 years from generation III to generation VI and an increase of the pathological repeat size from a minimum of 51 to a maximum of 67 repeats, respectively.
Fig. 3
Anticipation in SCA4 in subsequent generations of families I and II and inverse correlation between age of onset (AOO) and repeat length (RL) of 26 SCA4 patients
Furthermore, nine non-related patients (Tab. 2), with dominant ataxia, turned all out to be carriers of a ZFHX3 expanded allele where two patients carry expansions of only 44 and 45 GGC repeats.
Clinical characterizationThe clinical phenotype of the members of family I is described in the following case reports and in Table 1.
III 12 (55 repeats)This male patient first noticed an unsteady gait, slurred speech and swallowing problems at age 49 years. The symptoms progressed slowly and at the age of 58 he presented with marked dysarthria, areflexia, and severe gait ataxia. NCS confirmed an axonal polyneuropathy. The MRI revealed an atrophy of the cerebellum, medulla oblongata, and cervical spinal cord. The patient died of an unknown cause at the age of 59 years.
IV 8 (53 repeats)This female patient developed an unsteady gait at age 48 years with occasional twisting of her ankles. At the age of 54 years, examination revealed no dysarthria, but areflexia and gait ataxia. A CT scan showed mild cerebellar atrophy.
IV 10 (57 repeats)This female patient developed first symptoms at the age of 40 years. At the age of 82, she presented to the hospital due to a stroke. Regardless of the stroke she showed dysarthria, limb ataxia and a depressive mood.
IV 19 (53 repeats)This male patient developed dysarthria at the age of 48 years followed by progressive cerebellar ataxia with an inability to stand unaided at the age of 61 years. Aged 63, he presented with difficulties to sit without losing balance and falls from the wheelchair.
IV 25 (63 repeats)This male patient noticed an unsteady gait at age 25 years as well as mild slurring of his speech. At age 27, he showed mild dysarthria and mild cervical dystonia. The MRI showed distinct cerebellar atrophy. Gait ataxia progressed over time and the patient suffered from frequent falls. He started to use a walker at around the age of 32 years. Dysarthria was also progressive as well as tingling in finger and hands. At the examination at age 33 years, dysarthria and gait as well as limb ataxia have continuously progressed. For videos of patient see Online Resource 2 (Video 1).
V 6 (55 repeats)This male patient noticed an unsteady gait at the age of 48 years. At age 51 years, he presented with slow horizontal saccades, mild dysarthria, limb and gait ataxia, as well as areflexia. An MRI showed cerebellar atrophy and NCS confirmed sensory polyneuropathy of the upper and lower extremities independent of the nerve length.
V 16 (54 repeats)This male patient noticed an unsteady gait at the age of 50 years with swaying to both sides as well as reduced fine motor skills. At age 53 years, he presented with lower limb and gait ataxia, reduced vibration sense and areflexia. His gait ataxia progressed over the next 1.5 years to the need to use a walker outside the home. Clinical examination at the age of 54 years revealed progressive limb and gait ataxia, but still no dysarthria.
V 17 (65 repeats)This male patient noticed reduced sensation at the finger tips and feet at the age of 18 years. He developed an unsteady gait and slurred speech. Aged 27, he started urinary self-catheterization due to bladder emptying disorder. At the examination at age 29 years, he presented with slow horizontal saccades, dysarthria, gait ataxia and sensory neuropathy. An MRI showed atrophy of the cerebellum and NCS confirmed severe sensory neuropathy. All symptoms progressed over time and the patient additionally suffered from orthostatic hypotension. At around age 40, he started using a wheelchair. Aged 42 years, he presented with severe dysarthria, oromandibular dystonia, ocular apraxia with head movements while generating saccades. Ataxia had also significantly worsened.
V 18 (59 repeats)This male patient noticed first symptoms aged 18 years. At the age of 29 years, he presented with gait ataxia, areflexia, as well as hypesthesia and impaired proprioception in the lower limbs. He developed a progressive gait disorder with subsequently frequent falls, dysarthria and bladder emptying problems. When he was admitted to a hospital due a severe cervical spinal stenosis at the age of 47 years, he showed gait and limb ataxia, hypesthesia of both legs as well as reduced sense of vibration and proprioception. Although the neurosurgical operation was successful, he developed acute cardiac failure and pneumonia leading to death at age 47 years.
VI 3 (67 repeats)This male patient noticed an unsteady gait at age 21 years, followed by an impairment of his fine motor skills. At around 22 years of age, he noticed a slurred speech. At 24 years, bladder emptying disorder was diagnosed. Three years later, he presented with mild dysarthria, saccadic smooth pursuit, mild ataxia and areflexia at the lower limbs, and an unsteady gait. NCS revealed sensory neuropathy. An MRI demonstrated cerebellar atrophy without structural abnormalities of the spinal cord. Upon his examination at the age of 26 years, limb ataxia was now also present in the upper limbs and the patient showed slowed saccades. Gait ataxia progressed over the next 2 years with occasional falls, while dysarthria did not worsen. At the age of 36, he developed dysphagia which was rapidly progressive thereafter leading to food aspiration and subsequent pneumonia with the need manual ventilation. Diagnostic workup revealed atony and lack of motility of the esophagus. After restriction of further invasive therapy, the patient died at age 36 years. For video of the patient, see Online Resource 3 (Video 2).
Only limited information on patients IV 5 and V 8 was available (see Table 1).
In summary, patients initially manifested with a gait disorder due to a combination of cerebellar ataxia and sensory neuropathy. Both cerebellar ataxia and sensory neuropathy seem to have equally contributed to the phenotype and impairment of patients’ daily life. Autonomic dysfunction occurred only later in the disease and was clinically relevant only in patients with a severe manifestation. Disease progressed noticeable over a short amount of time, especially in patients with longer repeats, sometimes resulting in premature death of the patient.
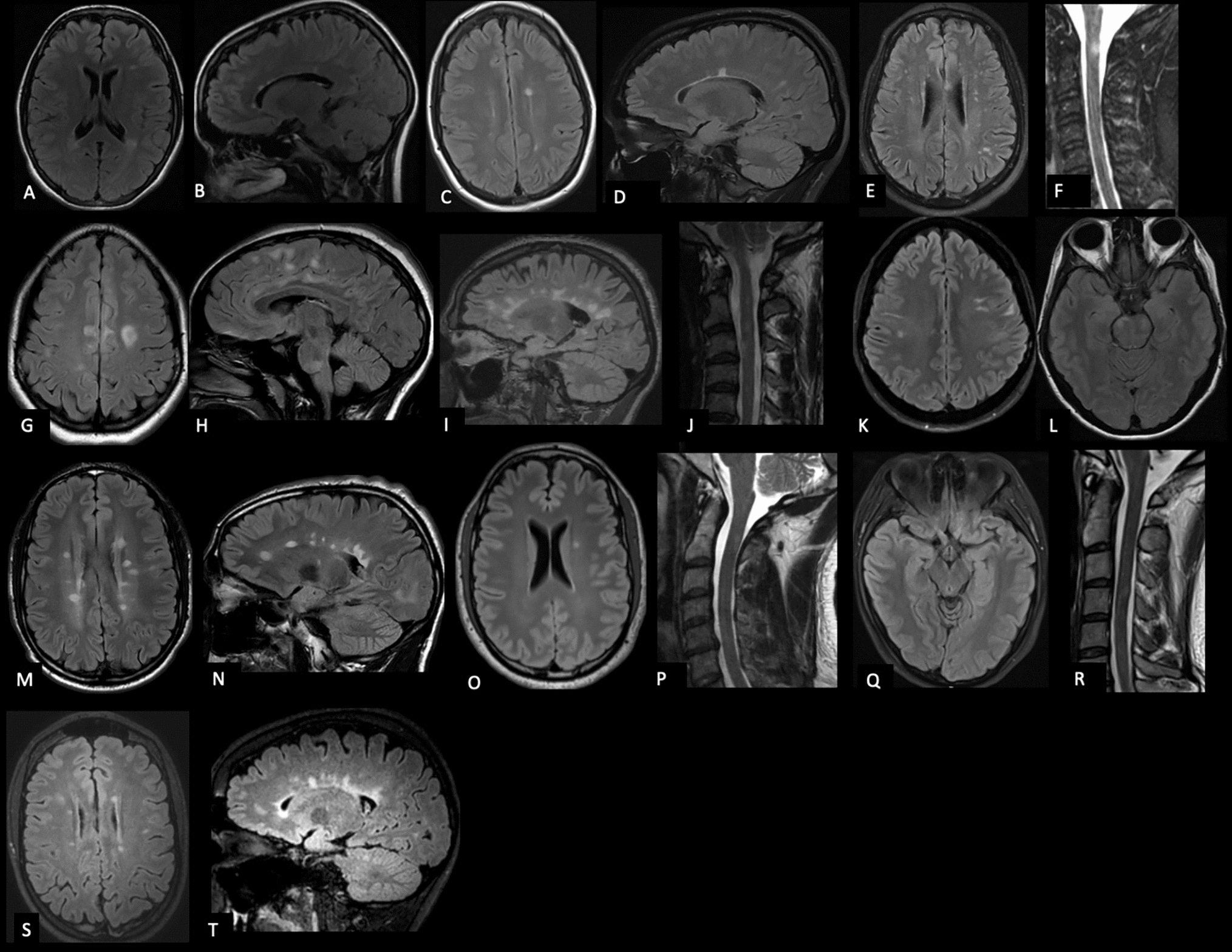
MRI analysisCerebellar volumetry revealed widespread gray matter atrophy in the two symptomatic ZFHX3 variant carriers but no significant atrophy in the presymptomatic carrier four years before his symptom onset (Online Resource 1, Fig. 2). Atrophy was present in most parts of the cerebellum including not only part of sensorimotor regions (lobules VI and VIII a/b) but also lobules associated with higher tasks (lobule VI and Crus I (language and verbal working memory), lobule VI (spatial tasks), lobules VI, VIIb and Crus I (executive functions) and lobule VI and Crus I (emotional processing). The anterior lobe (lobules I–V—sensorimotor tasks) did not show atrophy [12]. Neither did neurodegeneration affect cerebellar white matter in the investigated patients.
留言 (0)