
記住我



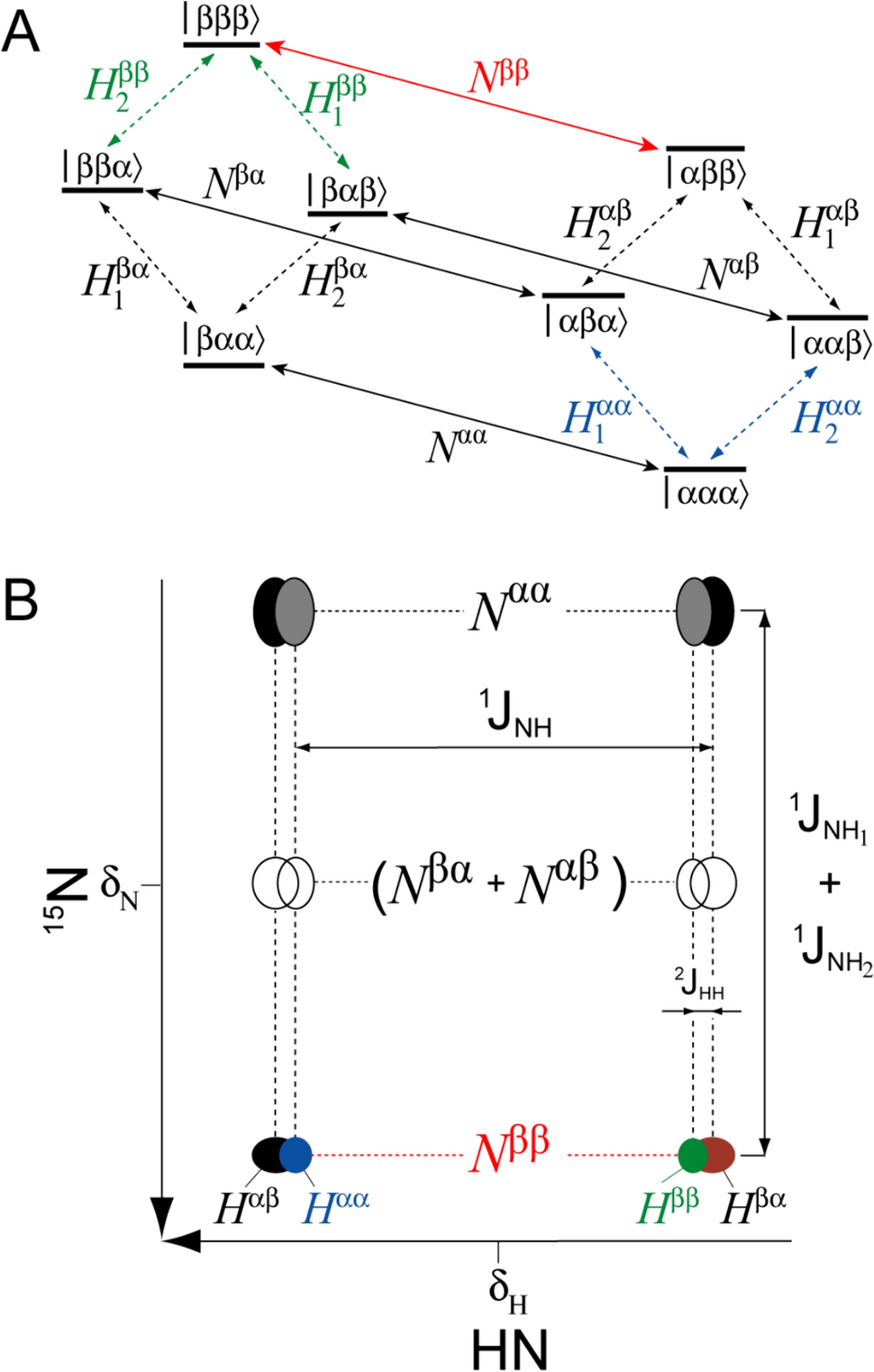


Figure 1A shows the energy level diagram of an isolated AMX (15N1H11H2) spin system (NH2 group). Each eigenstate is defined by three symbols, ‘α’ or ‘β’, where the first symbol corresponds to the state of the 15N spin, and the last two symbols, to the spin states of the two hydrogens, H1 and H2. The 2D multiplet pattern observed for an individual 1H spin of an 15N1H11H2 spin-system in a fully coupled HSQC experiment, is schematically shown in Fig. 1B, with the differences in peak widths intentionally exaggerated for illustration purposes. Among the four 15N transitions, the two central ones (Nαβ and Nβα) are not observable in the limit where the one-bond 15N–H J couplings, 1JNH1 and 1JNH2, are equal. Although differences in 1JNH of up to ~3 Hz have been reported for NH2 groups in model compounds (Bystrov 1976; McIntosh et al. 1997) — 1JNH = − 93.2 ± 1.3 Hz between 15N and the E proton (anti to carbonyl oxygen) and 1JNH = − 90.2 ± 0.9 Hz between 15N and the Z proton (syn to carbonyl oxygen) — these differences are predicted to be on a par with the linewidths of the central 15N transitions only for small proteins. In the 1H (horizontal) dimension, the multiplet pattern consists of a doublet of doublets, with the geminal two-bond 1H–1H J couplings, 2JHH, in NH2 groups of carboxamides small and positive (+ 2.3 ± 0.2 Hz in formamide (Chuck et al. 1969) and + 2.9 ± 0.5 Hz measured for 8 Asn/Gln NH2 groups in ubiquitin (Permi et al. 1999)). As 2JHH is in practice always much smaller than the difference in chemical shifts between the protons E and Z, no strong coupling effects are expected for NH2 groups under any circumstances. Below we formulate the theoretical basis for differential relaxation of 15N and 1H transitions in an 15N–1H2 spin-system and identify 15N and 1H transitions associated with the slowest transverse relaxation rates (shown in red and green/blue in Fig. 1 for 15N and 1H, respectively).
Fig. 1
NMR transitions in an 15N–1H2 spin-system. A Energy level diagram of an isolated AMX (15N1H11H2) spin system (NH2 group). Three symbols, ‘α’ or ‘β’, label each eigenstate, with the first symbol corresponding to the state of 15N, and the last two symbols, to the spin states of hydrogens H1 and H2. Diagonal solid arrows depict 15N transitions, each labeled with the single-transition operator defined in Eq. 1 and the Supplementary Information, SI, Eq. S1. Diagonal dashed arrows depict 1H transitions labeled with single-transition operators defined in Eq. 4 and SI, Eq. S4. The 15N transition associated with the slowest transverse relaxation rate and selected for in NH2-TROSY experiments is colored in red. The 1H transitions usually associated with the slowest transverse relaxation rates are colored in green, while those that can occasionally be associated with the slowest rates, are shown in blue. B Schematic representation of the 2D multiplet pattern observed for an individual 1H spin of an 15N–1H2 spin-system in the HSQC experiment performed without de-coupling in either dimension. The pattern is drawn for 1JNH < 0, 2JHH > 0, and 1JNH1 = 1JNH2. For 1JNH1 = 1JNH2, the intensities of 15N transitions Nαβ and Nβα vanish (shown with open circles). The frequencies of 15N and 1H nuclei are labeled with δN and δH, respectively. Horizontal, short-dashed and vertical, long-dashed lines depict 15N and 1H transitions, respectively. The transverse relaxation rate of each 15N transition is defined in Eqs. 2, 3 and SI, Eqs. S2–S3, while that of each 1H transition, in Eqs. 5, 6 and SI, Eqs. S5–S6. The 15N transition associated with the slowest transverse relaxation rate is colored in red, while the 1H peaks usually (Hββ) or incidentally (Hαα) associated with the slowest transverse relaxation rate (highest intensity) are colored in green and blue, respectively (see text) The outer 1H transitions, Hαβ and Hβα, are shown in black and brown, respectively
Each of the four 15N transitions of an NH2 spin-system (Fig. 1A) can be described by a single-transition operator, NPQ, representing a combination of product operator terms (Sorensen et al. 1983),
$$N^ = N_ + p \times 2N_ H_ + q \times 2N_ H_ + pq \times 4N_ H_ H_$$
(1)
where P and Q denote the eigenstates of the two protons HZ,1 and HZ,2, respectively: (P,Q) ∈ ; HZ = (Hα–Hβ)/2; and the multiplicative factors p and q are equal to + 1 and -1 for (P,Q) in the α and β states, respectively: (p,q) ∈ (see Supplementary Information, SI, for expansion of Eq. 1 into the vector \(\vec\) of all 4 15N components, Eq. S1). The cumulative rate of decay of transverse 15N magnetization in an isolated 15N–1H2 spin-system, R(NPQ), is composed of the following combination of transverse relaxation rates,
$$R(N^ ) = R_}_ }}^}}} + R_}_ }}^}}} + R_}}^}}} + pq \times R_}_ }_ }}^}} + p \times R_}_ }}^}} + q \times R_}_ }}^}}$$
(2)
where \(R_}}^\) and \(R_}}^\) are auto-and cross-correlated relaxation matrices for nuclei A(M), respectively, due to relaxation mechanisms j, k ∈ . The first two terms in Eq. 2 (\(R_}_ }}^}}} ,_} R_}_ }}^}}}\)) account for relaxation due to dipolar interactions of 15N with the two 1H spins; the third term (\(R_}}^}}}\)) describes relaxation due to 15N CSA; the fourth term (\(R_}_ }_ }}^}}\)) accounts for cross-correlated relaxation between the two dipoles (N–H1 and N–H2); and the last two terms (\(R_}_ }}^}} ,R_}_ }}^}}\)) account for cross-correlated relaxation between 15N CSA and the two N–H dipoles. In the macromolecular limit (with only the contributions from the spectral density function J at zero frequency, J(0), taken into account), and assuming an axially symmetric 15N CSA tensor, these rates are given by,
$$R_}_ }}^}}} = R_}_ }}^}}} = \frac\left( }}}^}}} } \right)^ S^ \tau_$$
(3a)
$$R_}}^}}} = \frac\left( }}^}}} } \right)^ S^ \tau_$$
(3b)
$$R_ NH_ }}^ = \frac\left( }}}^}}} } \right)^ P_ \left( }_ }_ }}^}} } \right)S^ \tau_$$
(3c)
$$R_}_ }}^}} = \frack_}}^}}} k_}}}^}}} P_ \left( }_ }}^}} } \right)S^ \tau_$$
(3d)
$$R_}_ }}^}} = \frack_}}^}}} k_}}}^}}} S^ \tau_ P_ \left( }_ }}^}} } \right)S^ \tau_$$
(3e)
where \(k_^ = - \left( /4\pi } \right)\hbar \gamma_^} \gamma_^} r_^\); \(k_}}^}}} = \gamma_}}^} \Delta \sigma_}} B_\); μ0 is the vacuum permeability constant; γi, the gyromagnetic ratio of spin i; rNH, the N–H inter-nuclear distance (1.02 Å); τC, the global molecular rotational correlation time (assumed isotropic); S, the generalized order parameter (assumed the same for all types of interactions); ΔσN, 15N CSA, Δσ = σZZ–(σXX + σYY)/2 (see ‘Materials and Methods’); B0, the static magnetic field; and the second-order Legendre polynomial, P2(cos(θμ,ν)) = (1/2)[3cos2(θμ,ν)–1], where θμ,ν is the angle formed between the principal (ZZ) axes of tensorial interactions μ and ν. The angle \(\theta_}_ }_ }}^}}\) = 118º was used in all calculations. Equation 2 does not include contributions to relaxation from 1H–1H dipolar interactions within NH2 groups (affecting only the central transitions of the NH2 multiplet) as well as dipolar interactions with 1H spins in the environment (protons ‘external’ to the NH2 group in question). The full treatment is provided in the SI, Eqs. S2–S3.
The four transitions of each 1H spin (Fig. 1A; H1 is singled out below) are described by,
$$H^ = H_ - p \times 2H_ N_ + q \times 2H_ H_ - pq \times 4H_ N_ H_$$
(4)
where P and Q describe the eigenstates of 15N and H2, respectively; NZ is defined as (Nβ–Nα)/2; and the same convention for (P,Q) and (p,q) is used as in Eq. 1 (see SI for expansion of Eq. 4 into the vector \(\vec_}}\) of all 4 1H components, Eq. S4). The cumulative rate of decay of transverse 1H magnetization in an isolated NH2 group, R(HPQ), is comprised of the following contributions,
$$R(H^ ) = R_}}}^}}} + R_}}}^}}} + R_}}^}}} + pq \times R_}}^}} + p \times R_}}^}} + q \times R_}}^}}$$
(5)
The first two terms in Eq. 5, \(R_}}}^}}}\) and \(R_}}}^}}}\), describe auto-relaxation due to dipolar interactions of a given 1H spin with 15N and another 1H spin, respectively; the third term (\(R_}}^}}}\)) accounts for auto-relaxation due to 1H CSA; the fourth term (\(R_}}^}}\)) accounts for cross-correlated relaxation between the H–N and H–H dipoles; and the last two terms (\(R_}}^}}\), \(R_}}^}}\)) account for cross-correlated relaxation between 1H CSA and H–N or H–H dipoles, respectively. In the macromolecular limit, and assuming an axially symmetric 1H CSA tensor, these rates are given by,
$$R_}}}^}}} = \frac\left( }}}^}}} } \right)^ S^ \tau_$$
(6a)
$$R_}}}^}}} = \frac\left( }}}^}}} } \right)^ S^ \tau_$$
(6b)
$$R_}}^}}} = \frac\left( }}^}}} } \right)^ S^ \tau_$$
(6c)
$$R_}}^}} = \frac\left( }}}^}}} } \right)\left( }}}^}}} } \right)P_ \left( }}^}} } \right)S^ \tau_$$
(6d)
$$R_}}^}} = \frack_}}^}}} k_}}}^}}} P_ \left( }}^}} } \right)S^ \tau_$$
(6e)
$$R_}}^}} = \frack_}}^}}} k_}}}^}}} P_ \left( }}^}} } \right)S^ \tau_$$
(6f)
where in addition to the definitions for Eq. 3: \(k_^ = k_^\); \(k_^ = - \left( /4\pi } \right)\hbar \gamma_}}^ r_}}}^\); rHH is the 1H–1H inter-nuclear distance (1.74 Å); \(k_}}^}}} = \gamma_}}^} \Delta \sigma_}} B_\), where ΔσH is the 1H CSA. The angle \(\theta_}}^}}\) = 31º was used in all calculations. Equation 5 does not include contributions to 1H relaxation from dipolar interactions with external 1H spins—see SI, Eqs. S5-S6, for the full treatment.
To gain quantitative insight into the magnitudes of 15N and 1H CSA (Δσ) and the angular terms in expressions for the CSA/DD cross correlated relaxation rates in Eqs. 3d–e and 6e–f, we performed DFT calculations of 15N and 1H CSA tensors in carboxamide NH2 groups (see ‘Materials and Methods’ and SI, Tables S1-S2). Figures 2A and B show typical orientations of 15N and 1H CSA tensors, respectively, relative to the carboxamide moiety. The calculated 15N Δσ is ~ −152 ppm, and is not significantly affected by protein environment (hydrogen bonding). The ZZ axis of the 15N CSA tensor lies practically (to within ~3º) in the carboxamide plane and forms an angle of ~20º with the bond vector connecting 15N with the E hydrogen, N-HE (the angle \(\theta_}_ }}^}}\) in Eq. 3d, labeled as ‘αN’ in Fig. 2A), and an angle of ~ 140º (or 40º since P2[cos(θ)] = P2[cos(180º-θ)]) with the bond vector connecting 15N with the Z hydrogen, N–HZ (the angle \(\theta_}_ }}^}}\) in Eq. 3e, labeled as ‘βN’ in Fig. 2A). The XX axis of the 15N CSA tensor is almost collinear (to within ~10–12º) with the N–C′ bond, while the YY axis is aligned with the normal to the carboxamide plane to within ~3–5º (Fig. 2A). These results (see typical values in the SI, Table S1) are in good agreement with earlier calculations of 15N CSA tensors in carboxamide NH2 groups of Asn/Gln side chains by Ernst and co-workers (Scheurer et al. 1999).
Fig. 2
Typical orientations of the CSA tensors of (A) 15N nuclei, and (B) 1HZ and 1HE nuclei in carboxamide NH2 groups. The directions of N–H bond vectors and the vectors connecting the two 1H nuclei are extended with short dashed lines. The angles \(\theta_}_ }}^}}\) and \(\theta_}_ }}^}}\) in Eqs. 3d–e are labeled by ‘αN’ and ‘βN’, respectively, in (A). The angles \(\theta_}}^}}\) and \(\theta_}}^}}\) in Eqs. 6e–f are labeled by ‘αH’ and ‘βH’, respectively, in (B). See ‘Materials and Methods’ for details of DFT calculations and SI, Tables S1 and S2 for the principal values and orientations of the 15N and 1HE/1HZ CSA tensors, respectively, obtained from the calculation with the program Gaussian for non-hydrogen-bonded and hydrogen-bonded side chain NH2 group of Gln41 in the protein ubiquitin
The calculated 1H CSA tensors are more sensitive to hydrogen bonding: the ΔσH values of ~8 and ~11 ppm for the E and Z hydrogens, respectively, were obtained in the absence of hydrogen bonding, while for the hydrogen-bonded cases, ΔσH generally increases to ~12–15 ppm for both hydrogens (note that although the calculated 1H CSA tensors are usually significantly asymmetric, we assume axial symmetry for the purposes of this discussion—see SI for exact representation of 1H CSA used in all calculations of relaxation rates and Table S2 for typical 1HE/Z CSA values). The ZZ axes of the CSA tensors of HE and HZ lie almost in the carboxamide plane (typically, within up to ~12º) forming the angles \(\theta_}}^}}\) (labeled with ‘αH’ in Fig. 2B) of ~20 ± 12º with respect to their respective H-N bond vectors, while the angles between the ZZ axes and the H–H vectors (\(\theta_}}^}}\); labeled with ‘βH’ in Fig. 2B) are larger, ~50 ± 12º. Note that from the geometry of the NH2 group, if the principal axis of the 1H CSA tensor was collinear with the N–H bond vector (i.e. \(\theta_}}^}}\)(αH) = 0), the angle \(\theta_}}^}}\)(βH) would be ~30º (Fig. 2B). It can also be noted from DFT calculations that both angles \(\theta_}}^}}\)(αH) and \(\theta_}}^}}\)(βH) tend to decrease by ~10–15º upon hydrogen bonding implying that hydrogen bonding reorients the ZZ axis of the 1H CSA tensor somewhat closer to the corresponding N–H bond vector.
Calculations of 15N relaxation rates in NH2 groups using Eqs. 2, 3 (see also SI, Eqs. S2-S3) show that NH1–NH2 dipole–dipole cross-correlated relaxation (\(R_}_ }_ }}^}}\), Eq. 3c) does not play a major role in 15N line narrowing, as the angle \(\theta_}_ }_ }}^}}\) is ~118º (P2[cos(118º)] = − 0.17), and the contributions of this cross-correlated relaxation mechanism are the same for the Nαα and Nββ transitions. The physical origin of line narrowing for the Nββ transitions (shown in red in Fig. 1) derives from the cross-correlated relaxation between 15N CSA and the two N–H dipoles (\(R_}_ }}^}}\) and \(R_}_ }}^}}\), Eqs. 3d–e), as is the case for 15N-1H spin-systems (Pervushin et al. 1997; Goldman 1984). However, the line-narrowing effects induced by the CSA/DD cross-correlated relaxation are substantially weaker in 15N–1H2 spin-systems compared to their 15N-1H counterparts for the simple reason that 15N nuclei are relaxed by dipolar interactions with two 1H spins (Eqs. 2 and 3a), while the ZZ axis of the 15N CSA tensor is approximately aligned only with the N-HE bond vector (angle αN in Fig. 2A; see discussion above and (Scheurer et al. 1999)). As a consequence, the CSA/DD cross correlated relaxation rates in Eqs. 3d–e differ by a factor of ~2 (for \(\theta_}_ }}^}}\)(αN) = 20º and \(\theta_}_ }}^}}\)(βN) = 40º, P2(αN)/P2(βN) = 2.2), and the relaxation due to the N-HZ dipolar interaction is not compensated for by the CSA/DD cross correlation to the same extent as that due to the N-HE dipole–dipole interaction. The field dependence of 15N TROSY linewidths for 15N–1H and 15N–1H2 spin-systems are compared in Fig. 3, showing that the optimal 15N TROSY effect for 15N–1H2 spin-systems is achieved at ~40% higher magnetic field strength than for their 15N–1H counterparts.
Fig. 3
Plots showing the dependence of 15N TROSY linewidths (15N-R2/π; Hz) on the strength of the static magnetic field, B0, expressed in units of 1H frequency (GHz) for isolated 15N–1H (black) and 15N–1H2 (red) spin-systems. The calculations were performed for the product S2τC equal to 25 ns using the angles \(\theta_}_ }}^}}\) = 20º and \(\theta_}_ }}^}}\) = 40º for 15N–1H2 spin-systems, and the angle \(\theta_}}^}}\) = 16º for 15N–1H spin-systems. 15N CSA (ΔσN) values of -152 ppm and -164 ppm for NH2 and NH groups, respectively, were used, along with the N–H internuclear distance rNH = 1.02 Å for both spin-systems. Note that the minimal 15N linewidths of NH2 groups (red) is ever so slightly narrower (by 2.5%) than that of their NH counterparts (black). This is a consequence of NH-NH dipole–dipole cross correlated relaxation in NH2 spin-systems, Eq. (3c). Although relatively small in magnitude (see text), these cross correlations lead to the same narrowing of the lines of both the TROSY (Nββ) and anti-TROSY (Nαα) 15N components in NH2 groups. When the contribution of NH–NH dipole–dipole cross correlated relaxation is excluded from the calculation, the minimal linewidth for NH2 groups becomes 2.6-fold broader than that of its NH counterpart (the red curve shifting upwards by ~2.7 Hz)
The effects of cross-correlated relaxation on the relative relaxation rates/intensities of the 1H transitions in 15N–1H2 spin-systems are more subtle. Unlike in 15N–1H spin-systems, the cross-correlated relaxation between the H–N and H–H dipoles (\(R_}}^}}\); Eq. 6d) is the main determinant of line narrowing of the Hαα and Hββ transitions in NH2 groups—the inner lines of the NH2 quadruplet, shown in blue and green, respectively, in Fig. 1. With the angle \(\theta_}}^}}\) = 30º, these cross-correlated relaxation rates have the same negative contributions to the rates of the Hαα and Hββ transitions (Eq. 5), that amount to ~1/2 of the auto-relaxation rates due to H–H dipolar interactions (\(R_}}}^}}}\)). The relative relaxation rates/intensities of the Hαα and Hββ transitions are dependent on the interplay between the 1H CSA/HN DD and 1H CSA/HH DD cross-correlated relaxation mechanisms (\(R_}}^}}\) and \(R_}}^}}\), Eqs. 6e–f). The calculated angles \(\theta_}}^}}\) ~20 ± 12º and \(\theta_}}^}}\) ~50 ± 12º (the angles αH and βH, respectively, in Fig. 2B), lead to the following relationships: (\(R_}}^}}\) < 0; \(R_}}^}}\) > 0; |\(R_}}^}}\)| >|\(R_}}^}}\)|) for the Hββ transitions, and (\(R_}}^}}\) > 0; \(R_}}^}}\) < 0; |\(R_}}^}}\)| >|\(R_}}^}}\)|) for the Hαα transitions. As a consequence, in the vast majority of NH21H multiplets inspected, the rates \(R_}}^}}\) dominate, leading to the narrowest (most intense) lines in the 1H dimension of 2D NMR spectra corresponding to the Hββ transitions (green in Fig. 1). However, a small number of exceptions when the transitions Hαα (blue in Fig. 1) and Hββ have the same intensities or the former is slightly higher, were noted in the NMR spectra, and can be associated with those cases where \(\theta_}}^}}\)(αH) < ~12º, \(\theta_}}^}}\)(βH) < ~ 38º, and |\(R_}}^}}\)| >|\(R_}}^}}\)|. These rare instances may tentatively be attributed to strong hydrogen bonds formed by NH2 hydrogen(s) with oxygen acceptors in carbonyl groups of the protein or water molecules.
Hindered rotation about the Cγ–N (Asn) and Cδ–Nε (Gln) bonds that interchange the two amide protons, will not affect the net results of the relaxation effects discussed here. Earlier NMR measurements performed at 35 ºC, yielded rotation rates in the range 0–1 s−1 for side chains buried in the protein core, and 1–10 s−1 for mobile side chains (Guenneugues et al. 1997). A more recent NMR study (Wang et al. 2021) showed, however, that the rotation rates are highly temperature dependent and decrease by ~3–~5-fold upon a temperature decrease from 35 to 15 ºC. Since TROSY effects are demonstrated here with experiments performed at 5 ºC, we do not expect that rotation rates on the order of 1–2 s−1 would influence any of the cross-correlated relaxation rates discussed above. We note that even in the case of anomalously high rotation rates at 5 ºC, the individual 15N CSA/N–H DD cross-correlated relaxation rates (\(R_}_ }}^}}\) and \(R_}_ }}^}}\) in Eqs. 3d–e) will be partially averaged without net effects on the relaxation rates of the Nαα or Nββ transitions. Likewise, as the principal values and orientations of the 1H CSA tensors for the protons E and Z are similar (see ‘Materials and Methods’), the bond rotation that inter-changes these two protons is not expected to significantly affect the relaxation rates of each individual 1H spin.
Experimental verificationThe pulse scheme for selection of each of the 8 NH2 multiplet components is shown in Fig. 4, and is based on the experiment developed for selection of 1H–13C multiplet components in 13CH2 groups by (Miclet et al. 2004). Table 1 lists the phase settings in the pulse scheme of Fig. 4 used for selection of each multiplet component. Following the transfer of magnetization to 15N, the element of duration 2τb (enclosed in a solid rectangle in Fig. 4) selects for the desired 15N component, with the phase ϕ2 controlling which component is isolated (Nαα or Nββ). Subsequently, the element of duration 8τb (enclosed in the dashed rectangle, Fig. 4) eliminates the signals from backbone N–H groups some of which would otherwise overlap with the Asn and Gln side chain NH2 correlations. Following the ZZ-filter (gradient g5), the chemical shift of the selected 15N component is evolved during the t1 acquisition period, and 15N magnetization is subsequently selectively transferred to one of the 4 1H components of the HE and HZ protons by a variation of the planar Total Correlation Spectroscopy (TOCSY) element (Schulte-Herbrüggen et al. 1991; Mádi et al. 1997). This planar TOCSY based transfer can be distinguished from the SPITZE-HSQC approach developed earlier for simultaneous measurements of 1DCH and 1DHH residual dipolar couplings in 13CH2 groups of proteins (Carlomagno et al. 2000).
Fig. 4
Pulse scheme for selection of each of the eight NH2 multiplet components. All narrow and wide rectangular pulses are applied with flip-angles of 90° and 180°, respectively, along the x-axis unless indicated otherwise. The 1H carrier is positioned at the water resonance, while the 15N carrier is placed at 110 ppm. All 15N pulses are applied with the highest possible power (RF field strength 6.25 kHz). The steady-state Boltzmann 15N polarization is added to the magnetization transferred from 1H by appropriate choice of the phase ϕ1. 13C decoupling is intended for U-[13C; 15N]-labeled samples and is achieved using 180º adiabatic WURST-20 pulses (Kupce and Freeman 1996) (2.5 ms; ± 30 kHz inversion bandwidth) in a p5m4 composite decoupling scheme (Tycko et al. 1985), with the 13C carrier placed at 110 ppm. These pulses are designed for simultaneous decoupling of carbonyl (13C′) and aliphatic (13Cα/β/γ) carbon regions. The shaped 1Hϕ1 pulse at the beginning of the scheme is a ~7 ms 90º water selective pulse applied with an EBURP-1 profile (Geen and Freeman 1991). To achieve good suppression of the water signal for any phase of the 1Hϕ6 pulse (2τ1 period), this pulse is flanked by two rectangular water-selective 1H pulses (shown with shaded rectangles) of ~1.5 ms duration applied with the phases x and -x, respectively. Delays are as follows: τa = 2.25 ms; τb = 1/(16JNH) = 690 μs; τ1 = 0.34/(2JNH) = 1.89 ms; τ2 = 0.23/(2JNH) = 1.28 ms; δ = 800 μs; ε = 500 μs. The phase cycling for ϕ1, ϕ2, ϕ6 and ϕ7 is as indicated in Table 1; ϕ3 = 2(x), 2(-x); ϕ4 = 2(x), 2(-x), 2(y), 2(-y); ϕ5 = x; receiver = x,-x,-x,x, -x,x,x,-x. The durations and strengths of the pulsed-field gradients applied along the z-axis in units of (ms; G/cm) are: g1 = (0.4; 35), g2 = (1.5; 40), g3 = (0.3; 35), g4 = (0.5; 35), g5 = (1.0; 35), g6 = (0.5; 30), g7 = (0.1; 35), g8 = (0.4; 35), g9 = (0.102; 30). In addition, each of the encoding and decoding gradients g6 and g9 are applied along the x and y axes with the strengths of ~15 G/cm. Quadrature detection in t1 is achieved using the Rance-Kay gradient selection scheme (Kay et al. 1992; Schleucher et al. 1993), with ϕ5 inverted together with the gradients g6 for each complex point in t1
Table 1 Phase Settings in the Pulse Scheme of Fig. 4 for Selection of Each of the NH2 Multiplet Componentsa)The phases of 180º 1H pulses ϕ6 and ϕ7 (see Table 1) control which 1H component is selected as described in detail by (Miclet et al. 2004). Briefly, when the component Nββ is isolated before the t1 period and it is assumed that 1J\(_}_ }}\) = 1J\(_}_ }}\) = 1JNH, between the time-points a and b of the pulse scheme in Fig. 4 with (ϕ6, ϕ7) = (x, x), the 15N magnetization is transferred to 1H according to the following transformation,
$$\begin - N_^ \mathop\limits^(H_^} - 4H_^} H_^} N_ )\left\ \tau_ ) + \cos (\pi J_ \tau_ )\sin (\pi J_ \tau_ )} \right\} + \hfill \\ (2H_^} N_ - 2H_^} H_^} )\left\ \cos (\pi J_ \tau_ )\sin (\pi J_ \tau_ )\cos (\pi J_ \tau_ ) + \hfill \\ [1 + \sin^ (\pi J_ \tau_ )]\cos (\pi J_ \tau_ )\sin (\pi J_ \tau_ ) \hfill \\ \end \right\} \hfill \\ \end$$
(7)
Durations of the delays τ1 and τ2 are found by equating the trigonometric terms in the curly brackets in Eq. 7 and maximizing their sum, to yield: τ1 = 0.34/(2JNH); τ2 = 0.23/(2JNH). The optimal efficiency of the Nββ → Hββ transfer f = 0.85 (Miclet et al. 2004), where 1 is the efficiency of the 15N → 1H transfer in the (fully decoupled) sensitivity-enhanced HSQC experiment optimized for 15N-1H2 spin-systems (Schleucher et al. 1994). It is important to note that the expression for the transformation in Eq. 7 holds only in the basis of spherical operators for 15N and 1H magnetization underscoring the importance of using pulsed field gradients for coherence selection.
The experiment in Fig. 4 was tested on the U-[15N]-labeled sample of the buried cavity mutant, L99A, of T4 lysozyme at 5 ºC (τC ~ 20 ns). Keeping in mind that comparison of sensitivities of the TROSY and decoupled HSQC spectra recorded with identical sampling (acquisition) times may be biased in favor of TROSY experiments (Miclet et al. 2004), we measured first the relaxation times of the 1H-decoupled 15N SQ coherences and the TROSY 15N component (Nββ) for 26 separated NH2 correlations of the T4 lysozyme mutant (5 ºC; 900 MHz). The average relaxation times, < T2 > , are 55 ± 19 ms and 130 ± 55 ms for the decoupled 15N coherences and Nββ components, respectively. This helped us to choose an experimentally realistic scenario where the maximal acquisition time in the indirect (15N) dimension, t1,max, is set to a value approximately ‘half-way’ between these two averages (t1,max = 88 ms was used in practice). This t1 acquisition time is on the one hand sufficient for resolving all NH2 correlations in the 2D spectra of the T4 lysozyme mutant, and on the other hand, is not excessively long to significantly bias the comparison of the two experiments.
Selected components of the NH2 multiplet obtained with the pulse scheme of Fig. 4 for the T4 lysozyme mutant (5 ºC; 900 MHz; t1, max = 88 ms) are shown in Fig. 5. The two multiplet components along the 15N dimension of the 2D spectra, corresponding to the 15N transitions Nαα and Nββ (both obtained with selection of the narrowest 1H transitions, Hββ, in the 1H dimension of the 2D spectra), are shown in black and red, respectively, in Fig. 5A. Significant line narrowing is observed for the Nββ components. While weaker than for 15N-1H spin-systems at the same B0 field (Fig. 3), this effect is strongly B0 field-dependent, as the physical origin of differential relaxation of the Nαα and Nββ transitions derives from destructive interference of 15N CSA and N–H dipolar mechanisms (Eqs. 3e–d). The 4 multiplet components along the 1H dimension of the 2D spectra, corresponding to 1H transitions HPQ, (P,Q) ∈ (all obtained with selection of the narrowest 15N transitions, Nββ, in the
留言 (0)