
記住我







We evaluated 279 cases of balanced chromosomal abnormalities and selected 191 cases with resolved breakpoints indicating a “simple” rearrangement (i.e., breakpoints at only two genomic locations and no significant genomic imbalance) for further analysis (Table S1). Using the most recent Human Gencode Reference, Release 45, GRCh38.p14 (Frankish et al. 2021), we then identified 66 cases in which at least one breakpoint overlapped a lncRNA (Table S2 and Table S3). Overall, 79 unique lncRNAs were directly disrupted in these cases, and four lncRNAs including MEF2C-AS1 and ENSG00000257522 were each disrupted in two unrelated individuals. In 30 of the cases, no genes of any other class aside from lncRNAs were directly disrupted by the breakpoints. In this report, we present seven cases disrupting five different lncRNAs as examples of the potential value of assessing lncRNAs as diagnostic etiologies (Table 1).
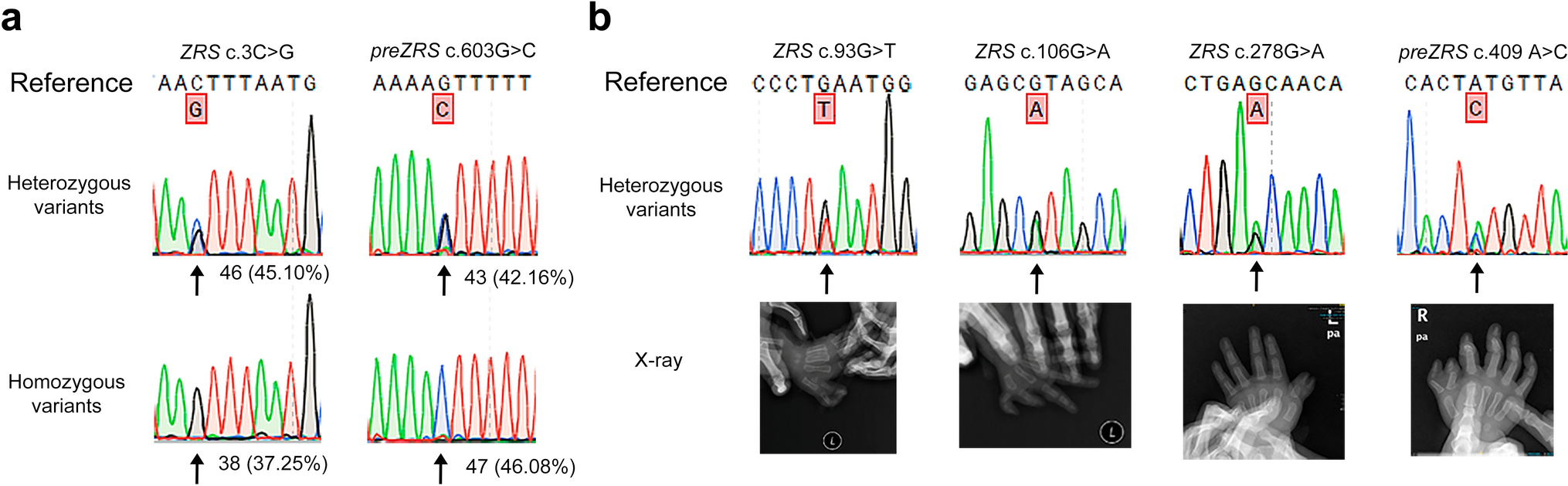
Table 1 Details regarding the breakpoints and the disrupted genes for the cases highlighted in this manuscript. Genomic coordinates refer to GRCh38/hg38 TBX2-AS1 is a candidate lncRNA for an association with hearing lossThe proband DGAP353 was diagnosed during gestation when her 24-year-old healthy mother underwent amniocentesis following an abnormal maternal serum screen for an elevated risk for trisomy 21. An apparently balanced translocation was detected in the female fetus between the long arms of chromosomes 14 and 17. Parental chromosome analyses revealed maternal inheritance and apparent structural identity to the maternal t(14;17) rearrangement. The daughter’s G-banded karyotype is described as 46,XX,t(14;17)(q24.3;q23)mat and the mother’s karyotype is 46,XX,t(14;17)(q24.3;q23). No clinical abnormalities were observed in the fetus and the pregnancy was continued. The daughter began developing signs of hearing loss at around the age of 10 years, and her hearing loss was found to be primarily sensorineural with a conductive element. By the age of 12 years, surgery was performed to rectify the conductive abnormalities, but the sensorineural hearing loss remained. The mother began wearing hearing aids at around the age of 40 years after a gradual decline in hearing for an unspecified time period. Both the daughter and her mother were otherwise healthy, typical of nonsyndromic deafness of unknown genetic etiology. Computerized tomography (CT) imaging of the temporal bones of the mother and daughter revealed abnormalities such as unusually small sinus tympani and narrowing of the round and oval windows (Fig. S1).
Both mother and daughter (DGAP353) harbor a translocation between chromosomes 14 and 17 with a 7 base-pair (bp) insertion of DNA of non-templated origin at the breakpoint in the der(17) chromosome (Fig. 1A). Following revision of suggested nomenclature (Ordulu et al. 2014), the next-generation cytogenetic nucleotide level research rearrangement is described below in a single string.
Fig. 1
The lncRNA TBX2-AS1 is a candidate for an association with hearing loss. (A) Chromosome diagrams depict the translocation between 14q23.3 and 17q23.2 in DGAP353. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 14q23.3 breakpoints in DGAP353. (C) Expanded view of the genomic region surrounding the 17q23.2 breakpoints in DGAP353. The directly disrupted lncRNA TBX2-AS1 is highlighted in red. ENSG00000267131 has been identified as an isoform of TBX2-AS1 by LNCipedia (Volders et al. 2019)
46,XX,t(14;17)(q24.3;q23)mat.seq[GRCh38] t(14;17)(14pter→14q23.3(+)(65,855,3)::17q23.2(+)(61,393,84)→17qter;17pter→17q23.2(+)(61,393,812)::TATATAC::14q23.3(+)(65,855,359)→14qter)mat
The DGAP353 breakpoints do not overlap any genes on chromosome 14 (Fig. 1B); however, this translocation results in the direct disruption of the lncRNA TBX2-AS1 from chromosome 17 (Fig. 1C). The Gencode annotation also lists the lncRNA ENSG00000267131 as a separate gene that is disrupted by these breakpoints, however this has been identified as an isoform of TBX2-AS1 (called TBX2-AS1:3) on the basis of shared exonic sequences by LNCipedia (Volders et al. 2019) (Fig. S2). While little is known regarding the biological role of TBX2-AS1, particularly in the context of hearing, the orthologous mouse lncRNA (2610027K06Rik) has been detected in the cochlear and vestibular sensory epithelium of embryonic and postnatal mice (identified as “XLOC_007930”) (Ushakov et al. 2017). Using the Gene Expression Analysis Resource (gEAR) portal (Orvis et al. 2021), we further found that while Tbx2-as1 is detected in supporting cell types (pillar and Deiters cells), it is predominantly expressed by sensory inner hair cells, as determined through cell-type-specific RNA-seq (Liu et al. 2018). Thus, the expression pattern of Tbx2-as1 is consistent with the finding that the hearing loss demonstrated by DGAP353 and her mother is primarily sensorineural.
The lncRNA gene TBX2-AS1 exists in a divergent configuration with the transcription factor TBX2 (MIM: 600747). Divergent lncRNAs have transcriptional start sites (TSSs) within 5 kb of another gene and are transcribed in the opposite direction in a “head-to-head” configuration. Importantly, the DGAP353 translocation does not disrupt the shared TBX2/TBX2-AS1 promoter region, but rather removes the 3’ end of TBX2-AS1, suggesting a potential lncRNA-dependent mechanism. Divergent lncRNAs have generally been associated with positive regulation of their neighboring gene, particularly when the neighbor is a transcription factor (Luo et al. 2016; Wang et al. 2020). Some divergent lncRNAs have also been found to modulate the downstream functions of the protein generated by the neighboring gene. Thus, knowledge regarding the function of the protein-coding member of a divergent pair can provide insights into the potential biological role of the lncRNA partner.
Intriguingly, TBX2 has previously been linked with hearing and inner ear development. In mice, Tbx2 has been associated with otocyst patterning in inner ear morphogenesis, as mouse models in which Tbx2 was conditionally knocked out exhibit cochlear hypoplasia (Kaiser et al. 2021). Previous studies have also shown that deletions encompassing TBX2 and TBX2-AS1 are found in individuals with hearing loss, albeit in conjunction with other deleted genes (Ballif et al. 2010; Nimmakayalu et al. 2011; Schönewolf-Greulich et al. 2011). In addition, a recent study has shown that Tbx2 is required for inner hair cell and outer hair cell differentiation, demonstrating that it is a master regulator of hair cell fate (García-Añoveros et al. 2022). Therefore, we suggest that the translocation disrupting TBX2-AS1 in DGAP353 and her mother may lead to altered expression or function of TBX2, ultimately resulting in the phenotype of hearing loss.
Recurrent disruptions of the lncRNA MEF2C-AS1 in individuals with neurological phenotypesWe additionally identified two cases, DGAP191 and DGAP218, with chromosomal rearrangements that disrupt the lncRNA MEF2C-AS1. The next-generation cytogenetic nucleotide level research rearrangements are described below in single strings.
DGAP191: 46,XY,t(5;7)(q14.3;q21.3)dn.seq[GRCh38] t(5;7)(5pter→5q14.3(+)(89,411,06)::7q21.3(+)(94,378,2)→7qter;7pter→7q21.3(+)(94,378,25)::5q14.3(+)(89,411,07)→5qter)dn
DGAP218: 46,XX,inv(5)(p12q13.1)dn.seq[GRCh38] inv(5)(pter→p14.2(+)(24,272,19)::q14.3(-)(89,105,02)→p14.2(-)(24,272,189)::TATTTATATGACAAG::q14.3(+)(89,105,031)→qter)dn
In both cases, the 5q14.3 breakpoints directly disrupt the lncRNA MEF2C-AS1. In DGAP191, the 7q21.3 breakpoints additionally overlap the lncRNA ENSG00000285090, but no protein-coding genes are directly disrupted (Fig. 2). In DGAP218, MEF2C-AS1 is the only gene of any class that is directly disrupted (Fig. 3).
Fig. 2
The lncRNA MEF2C-AS1 is disrupted in multiple individuals with neurological phenotypes, as shown here for DGAP191. (A) Chromosome diagrams depict the translocation between 5q14.3 and 7q21.3 in DGAP191. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 5q14.3 breakpoints in DGAP191. The directly disrupted lncRNA MEF2C-AS1 is highlighted in red. (C) Expanded view of the genomic region surrounding the 7q21.3 breakpoints in DGAP191. The directly disrupted lncRNA ENSG00000285090 is highlighted in red
Fig. 3
The lncRNA MEF2C-AS1 is disrupted in multiple individuals with neurological phenotypes, as shown here for DGAP218. (A) Chromosome diagrams depict the inversion between 5p14.2 and 5q14.3 in DGAP218. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 5p14.2 breakpoints in DGAP218. (C) Expanded view of the genomic region surrounding the 5q14.3 breakpoints in DGAP218. The directly disrupted lncRNA MEF2C-AS1 is highlighted in red
We previously reported both of these individuals as part of a larger set of cases with breakpoints in 5q14.3 (Redin et al. 2017). This region is of particular interest due to 5q14.3 microdeletion syndrome, which is characterized by neurological phenotypes including intellectual disability and epilepsy (Zweier and Rauch 2012). This syndrome is now recognized to be driven by decreased expression of MEF2C (MIM: 600662), either through direct disruption of MEF2C or due to distal mutations (Zweier and Rauch 2012). Indeed, when we previously described DGAP191 and DGAP218 (Redin et al. 2017), we noted that their phenotypes were similar to individuals with direct MEF2C disruptions. Furthermore, we determined that levels of MEF2C expression were reduced by ~ 30% in lymphoblastoid cell lines from both DGAP191 and DGAP218 (Redin et al. 2017); however, no mention was made of MEF2C-AS1. Recent studies have further elucidated the functional effects of altering MEF2C or its topological organization (Mohajeri et al. 2022), but the potential role of MEF2C-AS1 remains unclear.
While there is still little known regarding the function of MEF2C-AS1, it has recently been found that overexpression of MEF2C-AS1 can increase the levels of MEF2C in human cervical cancer cell lines by serving as a microRNA sponge (Guo et al. 2022). Interestingly, MEF2C-AS1 is transcribed through multiple putative enhancers of MEF2C (D’haene et al. 2019), providing another potential mechanism for this lncRNA to regulate expression of its neighboring gene, as has previously been described for lncRNAs such as Bendr (Engreitz et al. 2016) and Uph (Anderson et al. 2016). Thus, for DGAP191 and DGAP218 we now propose that the disruption of MEF2C-AS1 leads to decreased expression of MEF2C, resulting in neurological phenotypes.
The lncRNA ENSG00000257522 is recurrently disrupted in individuals with microcephalyOur analysis further identified two cases, DGAP245 and NIJ1, with chromosomal rearrangements that disrupt the lncRNA ENSG00000257522 (Figs. 4 and 5). These individuals exhibit shared phenotypes including microcephaly and defects of the corpus callosum (Table S1). The next-generation cytogenetic nucleotide level research rearrangements are described below in single strings.
Fig. 4
The lncRNA ENSG00000257522 is disrupted in multiple individuals with microcephaly, as shown here for DGAP245. (A) Chromosome diagrams depict the translocation between 3p22.2 and 14q12 in DGAP245. Above, large regions containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. The region shown surrounding 14q12 is a TAD, with its borders previously defined in (Dixon et al. 2012). No TAD was defined surrounding 3p22.2, so instead the region including 1 Mb on either side of the breakpoints is displayed. Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene (B) Expanded view of the genomic region surrounding the 3p22.2 breakpoints in DGAP245. (C) Expanded view of the genomic region surrounding the 14q12 breakpoints in DGAP245. The directly disrupted lncRNAs ENSG00000258028 and ENSG00000257522 are highlighted in red
Fig. 5
The lncRNA ENSG00000257522 is disrupted in multiple individuals with microcephaly, as shown here for NIJ1. (A) Chromosome diagrams depict the translocation between 8q21.13 and 14q12 in NIJ1. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the 8q21.13 breakpoints in NIJ1. (C) Expanded view of the genomic region surrounding the 14q12 breakpoints in NIJ1. The directly disrupted lncRNAs ENSG00000258028 and ENSG00000257522 are highlighted in red
DGAP245: 46,XY,t(3;14)(p23;q13)dn.seq[GRCh38] t(3:14)(3qter→3p22.2(-)(36,927,959)::CATTTGTTCAAATTTAGTTCAAATGA::14q12(+)(29,276,117)→14qter;14pter→14q12(+)(29,276,10)::3p22.2(-)(36,927,6)→3pter)dn
NIJ1: 46,XX,t(8;14)(q21.2;q12)dn.seq[GRCh38] t(8;14)(8pter→8q21.12(+)(78,898,16)::14q12(+)(29,296,33)→14qter;14pter→14q12(+)(29,296,328)::AAAT::8q21.12(+)(78,898,172)→8qter)dn
In both cases, the 14q12 breakpoints directly disrupt the lncRNA ENSG00000257522 as well as the overlapping antisense lncRNA ENSG00000258028. In DGAP245, the 3p22.2 breakpoints additionally disrupt the protein-coding gene TRANK1 (MIM: 619316) (Fig. 4B), however this gene is not predicted to be haploinsufficient (pHaplo = 0.29) (Collins et al. 2022) and it has not been implicated in any human phenotypes by OMIM. In NIJ1, the 8q21.12 breakpoints disrupt the lncRNA MITA1 (Fig. 5B). Given that the only shared disruptions between these cases are to the lncRNAs ENSG00000257522 and ENSG00000258028, we focused on these for further analysis.
Using the GTEx database (Lonsdale et al. 2013), we found that ENSG00000258028 is not readily detected in neural tissue, and thus it is unlikely to cause the patient phenotypes. In contrast, ENSG00000257522 is primarily expressed in neural tissue (Fig. S3A), suggesting that it could play an important neurological role. Moreover, ENSG00000257522 exists within the same TAD as the protein-coding gene FOXG1 (MIM: 164874), which similarly exhibits a predominantly neural expression pattern (Fig. S3B). Disruptions in FOXG1 have been associated with a variant of Rett syndrome (MIM: 613454) (Ariani et al. 2008) as well as FOXG1 syndrome (Kortüm et al. 2011). Core phenotypes of these syndromes include microcephaly and corpus callosum defects, implicating FOXG1 dysregulation as the underlying genetic etiology in DGAP245 and NIJ1. Thus, we sought to identify potential regulatory elements that could be disrupted by the chromosomal rearrangements in these cases, and found three regions with prominent H3K4me1 chromatin modification (Fig.S3C), which is associated with enhancer activity (ENCODE Project Consortium 2012). Notably, one of these regions also exhibited H3K27Ac modification, which is also associated with enhancer activity (ENCODE Project Consortium 2012). Furthermore, these three regions each include VISTA enhancers that have been demonstrated to drive reporter expression in neural tissue in vivo in transgenic mice (hs566, hs1539, and hs1168) (Visel et al. 2007), and thus these regions exert experimentally validated enhancer activity.
Strikingly, all three of these enhancers exist within the lncRNA ENSG00000257522. While the most distal enhancer is partially disrupted by the breakpoints in DGAP245, the other two enhancers remain in the appropriate position relative to FOXG1. In NIJ1, all three of the enhancers are proximal to the breakpoints and are not separated from FOXG1. Thus, these enhancers are not directly disrupted by the chromosomal rearrangements, and instead their activity could be impaired due to the disruption of the lncRNA in which they are embedded. Indeed, transcription of lncRNAs through enhancers is a well-documented mechanism through which lncRNAs can regulate gene expression (Statello et al. 2021). Thus, we propose that the lncRNA ENSG00000257522 regulates the expression of FOXG1 through its effects on the embedded enhancers. This is consistent with previous findings that several lncRNAs function to modulate the expression of transcription factors and that this tight regulation is essential for maintaining proper functions of the transcription factors, particularly for pioneer factors such as FOXG1 (Ferrer and Dimitrova 2024).
Further supporting this, we also identified an individual with a complex de novo rearrangement that similarly disrupts ENSG00000257522. This individual, DGAP246, exhibits consistent phenotypes including microcephaly (Redin et al. 2017). The complex rearrangement in DGAP246 consists of 14 pairs of breakpoints, including eight breakpoints in 14q12. Overall, this results in the direct disruption of the lncRNA ENSG00000257522 while leaving the two most proximal enhancer elements in their correct position relative to FOXG1 (Fig. S3C). Taken together, these three cases implicate the lncRNA ENSG00000257522 in the regulation of FOXG1. Additionally, previous studies have reported several individuals with FOXG1 syndrome that harbor disruptions in this region, including a translocation in “Patient 1” that directly disrupts ENSG00000257522 (Mehrjouy et al. 2018). Similarly, a recent case report described another individual with FOXG1 syndrome whose balanced translocation had breakpoints mapping within the ENSG00000257522 lncRNA (Craig et al. 2020). Thus, we propose that disruptions of this lncRNA can cause phenotypes including microcephaly and defects of the corpus callosum, consistent with FOXG1 syndrome.
Potential regulation of KIRREL3 by its neighboring lncRNA ENSG00000255087We previously described DGAP148 as an individual with a neurodevelopmental disorder including attention deficits and difficulty with spatial coordination (Talkowski et al. 2012b). We have recently received updated information from the referring clinical geneticist indicating that this individual is in overall good health but continues to receive medication for attention-deficit/hyperactivity disorder (ADHD). She was not able to complete regular high school, however she works as a helper in a veterinary clinic. While she lives with her father, she is autonomous for tasks of everyday living, including meals, laundry, exercise, and driving. She is also described as very sociable.
DGAP148 has a de novo translocation (Fig. 6A), and the next-generation cytogenetic nucleotide level research rearrangement is described below in a single string.
Fig. 6
Disruption of the lncRNA ENSG00000255087 was identified in an individual with a neurodevelopmental disorder. (A) Chromosome diagrams depict the translocation between Xp11.4 and 11q24.2 in DGAP148. Above, TADs containing the breakpoints are shown, with the breakpoint positions indicated by vertical orange bars and the edges of the region shown in vertical pink bars. TAD borders were defined in (Dixon et al. 2012). Triangular contact maps display micro-C data indicating chromatin conformation (Krietenstein et al. 2020). H3K4Me1 and H3K27Ac tracks depict enhancer-associated chromatin modifications (ENCODE Project Consortium 2012). The VISTA track shows experimentally validated enhancer elements (Visel et al. 2007). Protein-coding genes are shown in blue and non-coding genes in green, with a single isoform depicted per gene. (B) Expanded view of the genomic region surrounding the Xp11.4 breakpoints in DGAP148. (C) Expanded view of the genomic r
留言 (0)