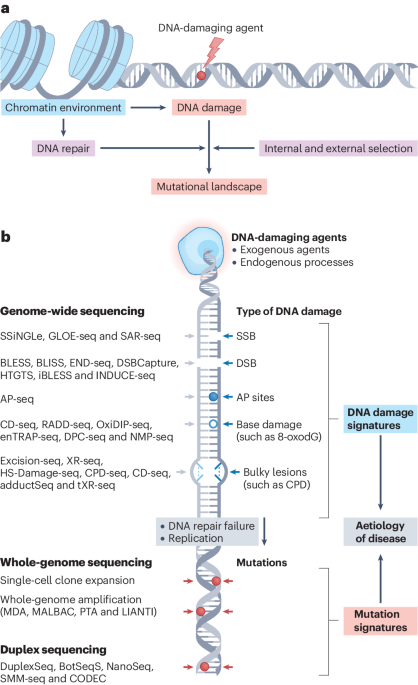
Decoding the language of chromatin modifications with MARCS
Most eukaryotic DNA is stored in the nucleus as chromatin, which provides a framework for the regulation of genome functions. Chromatin consists of histones and DNA, both of which carry various chemical modifications that orchestrate DNA-templated processes by directing nuclear proteins to their target genomic loci. Many nucleosomes bear multiple interconnected modifications that modulate local genome accessibility and serve as binding platforms for proteins that can ‘read’ these modifications. However, despite progress in identifying ‘readers’ of individual modifications, how the nuclear proteome decodes complex modification landscapes remains unclear.
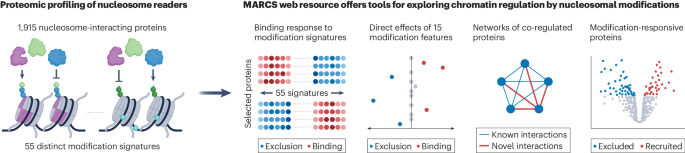
To address this question, we created a library of semi-synthetic dinucleosomes, incorporating modification signatures of promoter, enhancer and heterochromatin states. Using these dinucleosomes as baits in affinity purification experiments, we systematically examined the interactions between the human nuclear proteome and functionally distinct chromatin states by mass spectrometry1. We further developed computational tools for the analysis of this large-scale interaction proteomics dataset to quantitatively assess the direct effects of nucleosomal features on protein recruitment and exclusion. Using these tools, we identified networks of co-regulated factors, which connect chromatin modifications with downstream nuclear processes.



留言 (0)