
記住我

Ethics approval was received from The University of Queensland Human Research Ethics Committee (Approval No. HE2019002816).
This study focused on the optimisation and validation process for six qPCR assays using samples collected as per the protocol used for a multi-centre study comparing oral health of those with RTT with sibling controls. The multicentre study protocol is summarised herein to provide context to the downstream application of this proof-of-concept validation study. Briefly, the multi-centre study compared oral health status and oral health risk in individuals with RTT and sibling controls. Because of the location of treatment centres for RTT, this part of the study was multi-centre. The same clinician (YL) conducted dental examinations of individuals with RTT and siblings as controls at participants’ homes across the different sites. This part of the project included assessments of plaque factors that mediate the risk of oral diseases, the focus of this validation study.
Multicentre study sample populationThis consisted of those with RTT residing in Western Australia, South Australia and Queensland, registered with the population-based Australian Rett Syndrome Database (ARSD). Home visits occurred in January–February 2021 and November 2022 (South Australia), May 2022 (Western Australia) and June 2022 (Queensland). For the control population, medically healthy siblings who were living in the same household, or nearby suburb (if no siblings), without disability were recruited concurrently. Target recruitment numbers were based on approximately half the number of families residing within 1–1.5 h’ drive from the reference city centre, as indicated in Supplement 1 Table 1, with the total number of those with RTT (alive) and living in this area provided as reference. Considering geographical factors and the rarity of the disease, the conservative recruitment targets were considered reasonable. A short questionnaire was disseminated to inform a clinical examination. This was adapted from a previous version of a dental module of the InterRett database (Lai et al. 2022, 2021, 2023). Participants were contacted via telephone to discuss the study and any questions that arose before providing written consent.
Table 1 qPCR assay oligonucleotide and target informationData collectionAll participants’ families were contacted prior to each home visit. Selected additional participants also provided consent for taking of samples which were used for the assay optimisation stage which is the focus of this study.
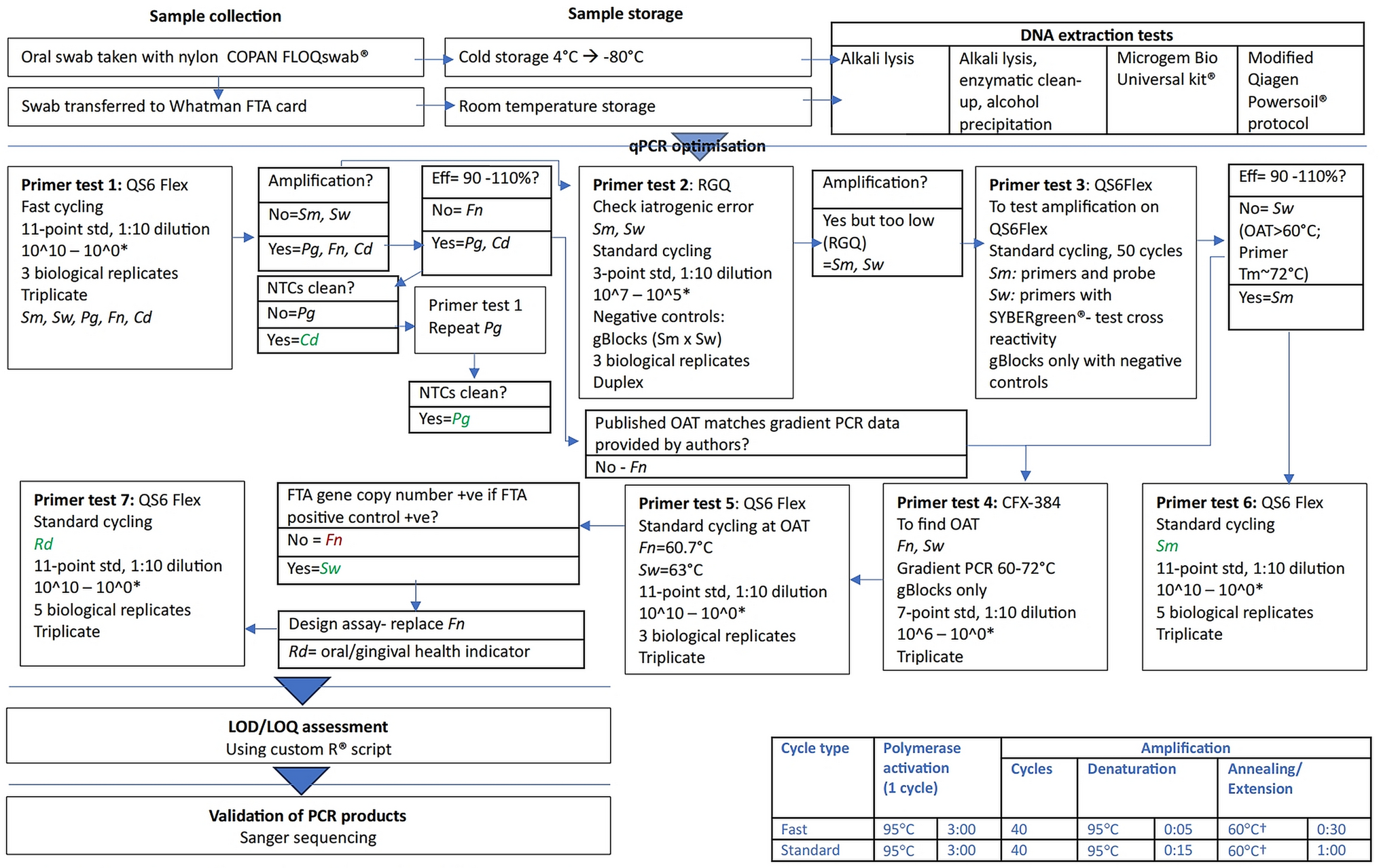
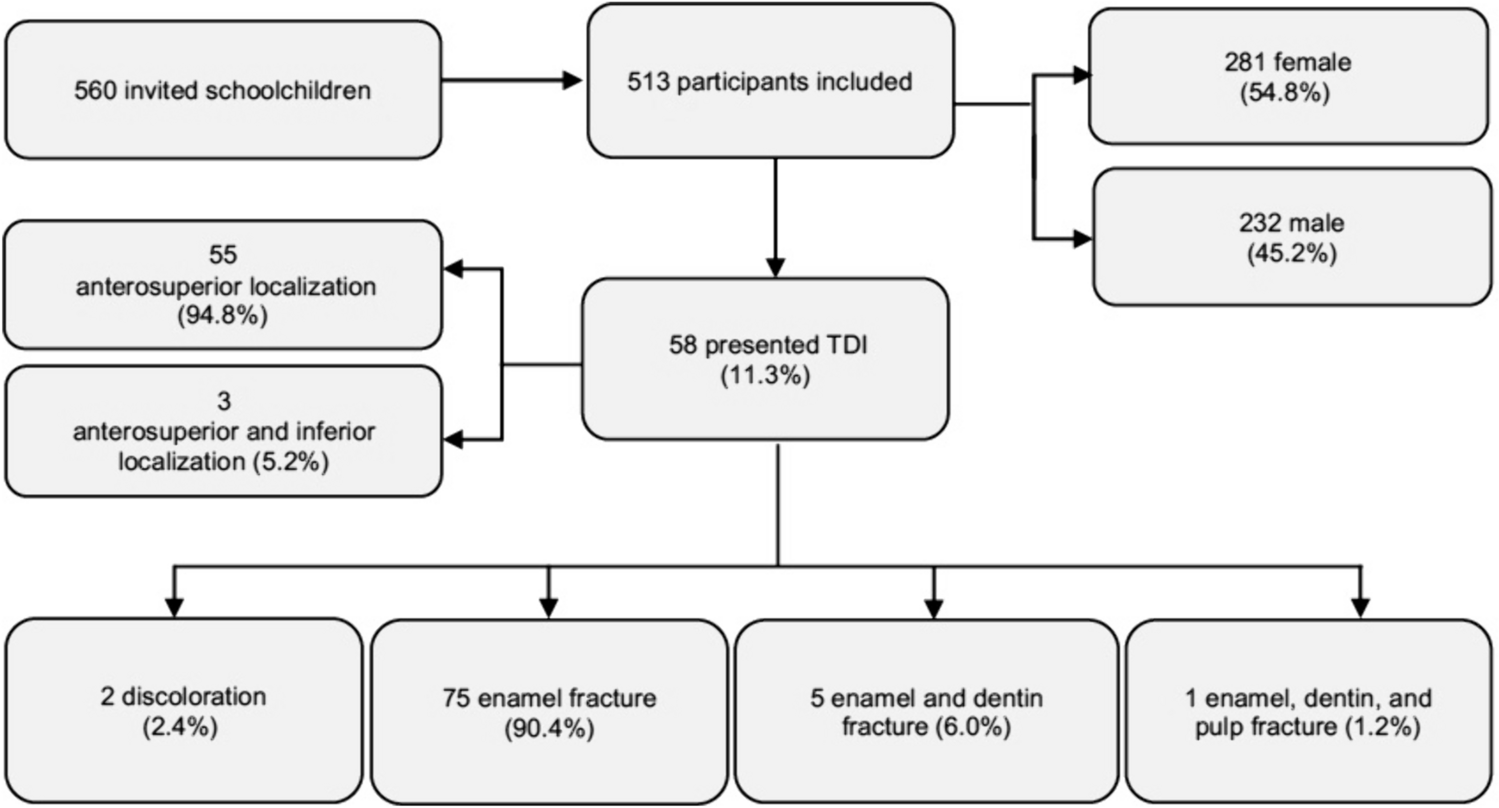
The methods described below related directly to this optimisation and validation study. The clinical workflow is illustrated in Fig. 1.
Fig. 1
Flow diagram illustrating workflow of proof-of-concept study. Legend: † Unless otherwise specified Cd = C. durum; CFX-384 = CFX-384 (Biorad): analysed with Bio-Rad CFX Maestro 1.0 Version 4.0.2325.0418 (Bio-Rad). Eff = qPCR efficiency; Fn = F. nucleatum subsp. fusiforme; LOD/LOQ = limit of detection/limit of quantification; NTC no template control; OAT optimal annealing temperature; PCR polymerase chain reaction; Pg = P. gingivalis; QS6 Flex Quantstudio 6 Flex; RGQ RotorGene Q 5-Plex HRM (Qiagen), analysis with Rotor-Gene Q Series Software v2.3.5 (Build 1) (Qiagen) with manual pipetting (not compatible with robotic pipetting).; Rd = Rothia dentocariosa/R. aeria; Sm = S. mutans; Std = standard; Sw = S. wiggsiae; *Shorthand notation for gene copy dilutions are based on the following calculated gene copies for bacterial targets at 10^10 gene copies: Cd = 12,833,183,536.50; Fn = 11,261,404,605.00; Pg = 19,198,587,102.00; Rd = 17,428,077,501.00; Sm = 18,903,502,168.50; Sw = 12,128,592,981.00
Dental plaque was collected for the assessment of microbiome dysbiosis that relates to the risk of dental caries (Philip et al. 2018). Briefly, supragingival dental plaque was collected with a sterile swab of the labial surface of the upper central teeth (or adjacent teeth if these were missing) and swabbed onto indicating archival storage cards (Micro FTA Indicating Cards, Whatman, Cat. No. WHATWB120211). The sterile swabs were placed in cold storage immediately, and transferred to − 20 °C storage within 4 h of data collection. Samples were later transferred to − 80 °C storage when practicable. The FTA cards were stored at room temperature, in line with manufacturer’s instructions.
Optimisation of DNA extraction processSelected samples were used to check both the quality and quantity of DNA from DNA extraction. Several DNA extraction protocols were tested, as described below.
Method 1. Modified Whatman extraction method b protocolA Qiagen Harris Unicore 2 mm micro-punch was used to isolate bacterial DNA for use in downstream extraction from classic FTA cards. Extractions were performed according to the Whatman protocol for alkali lysis from classic FTA cards Method B (Miles and Saul 2015) with modifications to the sample size and reagent volumes. The entire 30 mm diameter FTA samples were cut into small sections approximating 1 × 1 mm with a sterile surgical blade or surgical scissors that had been disinfected with 70% ethanol and DNAOUT™ DNA Removal Solution. The samples were washed with 600 µL of Qiagen FTA Wash solution (Cat # 2,719,978) and incubated for 5 min at room temperature (RT) with occasional mixing with an Eppendorf Thermostat C heating and cooling block heater. The supernatant was removed and washing procedure was repeated a further three times. The samples were then washed three times with 600 µL of TE buffer (10 mM Tris pH 8.0, 1 mM EDTA), and the tubes were incubated for a further 5 min at RT with occasional mixing and the liquid was removed with a pipette. The buffer was removed and 35 µL of alkaline incubation buffer added (0.1 N NaOH, 0.3 mM EDTA, pH 13.0). Samples were incubated at 65 °C (Method B) for 5 min. Then, 65 μL of neutralising solution (0.1 M Tris–HCl, pH 7.0) was added and the samples vortexed 5 times. Tubes were incubated for 10 min at RT. The tubes were then mixed by pulsed vortexing 10 times. The FTA samples were removed from the solution, ensuring that the maximum amount of elute was recovered.
Method 2. DNA extraction followed by enzymatic cleanup and ethanol precipitationDNA extraction was performed using the modified Whatman Extraction Method B outlined above, with additional repurification steps using Proteinase K and RNase A, followed by an ethanol precipitation step. Steps 2 and 3 of the New England Biolabs (NEB) 'Enzymatic Cleanup Protocol (removal of proteins and/or RNA) (New England Biolabs) were followed by an ethanol precipitation step to remove protein, RNA and residual chemical contaminants. Briefly, 1μL of Proteinase K and 1μL RNase A was added. Samples were mixed briefly and incubated at 56 °C for 5 min. For DNA ethanol precipitation, 0.1 vols 3 M sodium acetate was added to 2.5–3 vols ice cold 100% ethanol, and vortexed to mix thoroughly. Samples were precipitated at -20 °C overnight and centrifuged at full speed (14,000 rcf), 4 °C for 20 min. The pellet was washed twice with 0.5 mL ice cold 70% ethanol and centrifuged at 14,000 rcf at 4 °C for 10 min each time. The ethanol was removed by centrifuging quickly (10 s at 14,000 rcf). The pellet was air dried for 10 min and resuspended in 20 μL of nuclease-free water (1.000 g/mL at 3.98 °C (litre)). Finally, the pellet was incubated at 65 °C for 10 min with an Eppendorf Thermostat C heating and cooling block heater.
Method 3. DNA extraction using microgem bio Universal kitDNA extraction was performed at the Australian Genome Research Facility Brisbane laboratory, on one test sample (“YL FTA”) using the manufacturer protocol (Miles and Saul 2015). Briefly, 100 µL of water was added to 2 mm diameter punches. Samples were briefly vortexed and placed at room temperature for 15 min. The tubes were then vortexed and the liquid removed by pipette. Punches were resuspended in: 44 µL nuclease-free water, 5 µL MicroGEM BLUE Buffer and 1 µL prepGEM. Tubes were then placed in a thermal cycler and heated at 75 °C for 15 min followed by 95 °C for 5 min. Finally, liquid was decanted into tubes.
Method 4. DNA extraction using a modified Qiagen Powersoil Kit protocolDNA extraction was performed on a COPAN FLOQswab and its corresponding FTA card (approximately 2 mm punch), using the DNEasy Powersoil Pro kit (Qiagen) on an FTA sample and the corresponding swab sample.
DNA extraction was performed using a modified Powersoil protocol on samples labelled A19, B19 and C19, with the entire FTA sample included in the Powersoil reaction.
Preparation of the Streptococcus sanguinis culture was prepared as follows: 100 mL of Brain–Heart infusion (BHI) broth was prepared (3.7 g in 100 mL nuclease free water) and autoclaved at 121 °C. Then, 2 μL of S. sanguinis (ATCC 10556) culture was pipetted into 1 mL BHI broth and incubated for 48 h at 37 °C at 260 rcf on an Eppendorf Thermostat C heating and cooling block heater. The sample was centrifuged at 3900 rcf at 22 °C for 20 min to pellet the solution before discarding the supernatant. The pellet was resuspended in 100 μL nuclease-free water (1.0 g/mL at 3.98 °C (litre)). Then, half (50 μL) was pipetted onto an FTA card with sample label “B19” and allowed to sit for one hour. Samples B19 and C19 were then cut into small pieces approximating 1 × 1 mm with surgical scissors decontaminated with DNA-Out (Astral Scientific) followed by 70% ethanol, before including the entire samples in the Powersoil reaction. The other 50 μL of the S. sanguinis culture was then labelled as sample “A19”, and included directly into a Powersoil reaction.
In silico design of qPCR assaysThe study was conducted in line with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines (Bustin et al. 2009). Custom qPCR assays (primers and probes) targeting two caries-associated bacterial species (S. mutans, S. wiggsiae), one gingivitis-associated bacterial species (P. gingivalis) and 3 health-associated commensal bacterial species (F. nucleatum subsp. fusiforme, R. dentocariosa and C. durum) were designed using Primer3Plus and PrimerQuest (IDT). Specificity of the assays and off-targets were assessed using Nucleotide BLAST and Primer BLAST, while the IDT Oligoanalyzer Tool was used for assessing melting temperature and primer dimer profiles.
The in silico design process involved a comprehensive literature search for published species-specific assays where possible, with target amplicons verified by multiple sequence alignment using Multiple Alignment using Fast Fourier Transform (MAFFT) software; or else assays were custom designed using multiple sequence alignment using MAFFT software to identify species-specific sequences, and these formed the basis for synthetic DNA templates (gBlocks standards) design. Primers, probes, and synthetic DNA templates (gBlocks standards) were custom manufactured by Integrated DNA Technologies (IDT).
The qPCR oligonucleotide and target information are detailed in Table 1. The qPCR information on gBlocks is detailed in Table 2.
Table 2 qPCR assay gBlocks sequencesqPCR optimisationLysophilised samples were diluted according to manufacturer instructions. The qPCR protocol is summarised in Fig. 1. Primer (500 nM) and probe (250 nM) concentrations used the standard primer and probe concentrations recommended by the manufacturer (IDT), with a volume of 0.5 μL respectively. Primer and probe Mg2 + , and dNTP concentrations were 3 mM and 0.8 nM respectively. IDT PrimeTime Gene Expression Mastermix (IDT) was used with a volume of 5 μL, with 2 μL DNA template and 1.5 μL nuclease free water for a 10 μL reaction volume. Reaction setups were performed using an Assist Plus pipetting robot (Integra). An Applied Biosystems MicroAmp Optical 384-well reaction plate was used, in a Quantstudio 6 Flex (Applied Biosystems).
The DNA sample was mixed with an IDT Gene Expression (IDT) master mix, and robotically dispensed into a 384-well plate (10 μL/well, 2 µL DNA template/well). Synthetic DNA templates (gBlocks standards) served as the positive PCR control, and a non-template control was used to account for assay background. The gBlocks standards were run using a 1:10 dilution series from 10^0 to 10^10 dilutions on an 11-point standard curve, with the gBlocks concentrations shown in Fig. 1. Using test samples, optimised reactions were performed with the 384 well plate Quantstudio 6 Flex (Applied Biosystems). Assays were first run under the fast cycling conditions recommended by the manufacturer, and where assay efficiency was outside the range of 90–110%, assays were run under standard cycling conditions and gradient PCR was performed. Optimised assays were run with fast cycling conditions for C. durum and P. gingivalis assays, and on standard cycling conditions for S. mutans, and R. dentocariosa/R. aeria. The fast-cycling conditions were: enzymatic activation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 5 s, and 60 °C for 30 s. The standard-cycling conditions were enzymatic activation at 95 °C for 3 min, followed by 45 cycles of 95 °C for 15 s, and 60 °C for 1 min. Samples of S. wiggsiae and F. nucleatum subsp. fusiforme were run at standard cycling conditions, but with optimal annealing temperatures of 63 °C and 60.7 °C respectively, which were determined by running a gradient PCR using CFX-384 qPCR instrument (Biorad).
Data analysisData were analysed using Quantstudio software v1.2 (Applied Biosystems). The gene copy number in each sample was determined by comparing the CT values of each sample to those of the standards. Calibration curves for the primer tests were examined together with Cq (CT) variation, linear dynamic range, R2, and efficiency. The limit of detection (LOD) and limit of quantification (LOQ) were calculated using custom R scripts (Klymus et al. 2020). All assays were run in single plex.
Verification of qPCR productsPCR products for the assays were confirmed by Sanger sequencing using broth cultures containing the target species S. mutans (ATCC25175), F. nucleatum (subsp. 49,256), P. gingivalis (W50)) and test samples where they could not be sourced as a broth culture (S. wiggsiae, R. dentocariosa, C. durum).
The clinical workflow is illustrated in Fig. 1. For clarification for Primer Test 3, reactions were run in duplicate in 28 wells of 10 μL with setup as shown in Supplement 2 Table 1.
留言 (0)