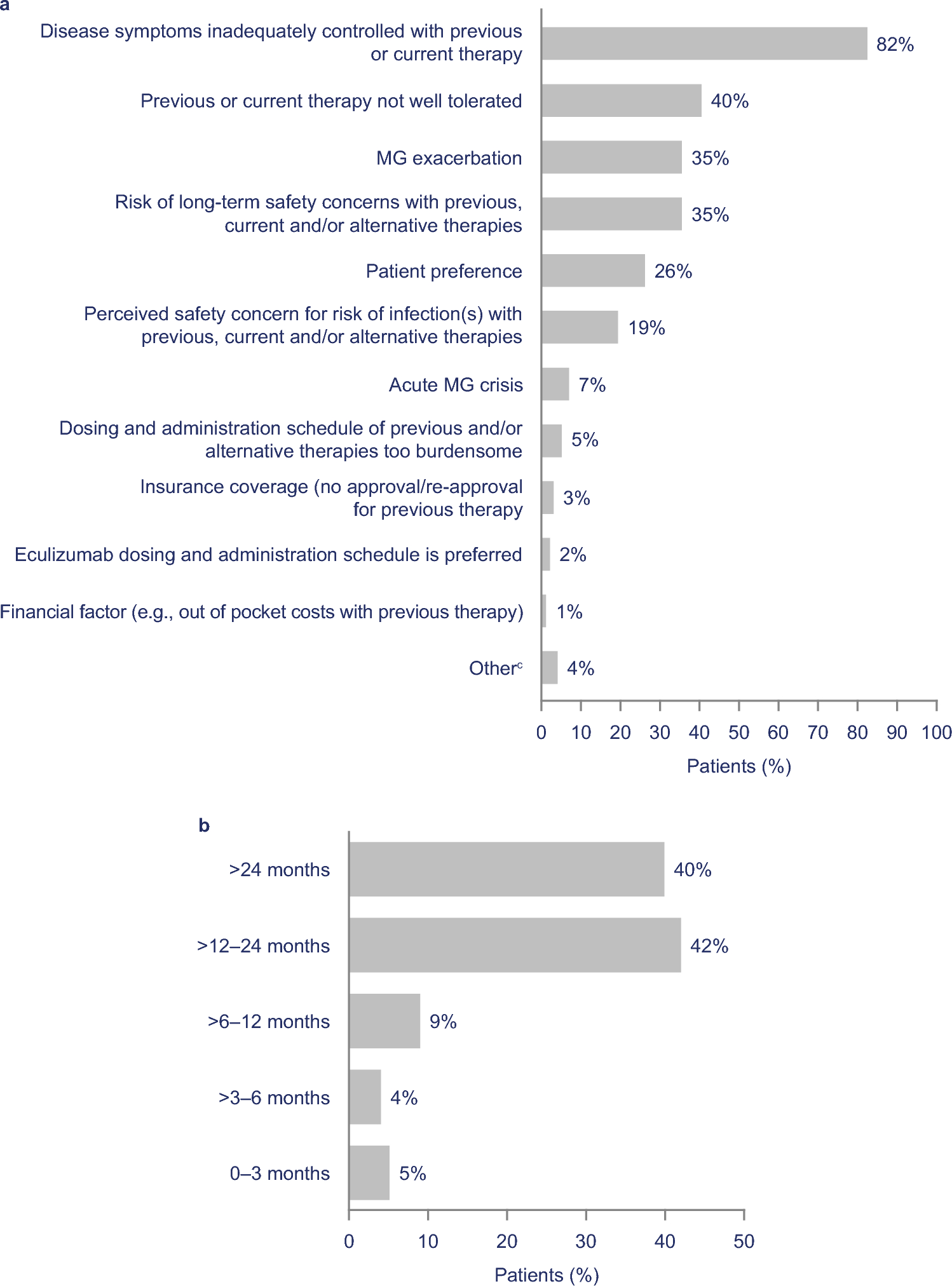
Prompted by the discovery of pathogenic biallelic repeat expansions in RFC1 in an ALS patient with additional sensory neuropathy and bilateral vestibular dysfunction [15], we investigated whether biallelic pathogenic repeat expansions in RFC1 could be another relevant genetic cause for motor neuron disease such as ALS, PLS or PMA. Therefore, we systematically characterized the RFC1 repeat array in a cohort of 107 patients with clinical diagnosis of MND (Fig. 1b, c, Supplementary Table 3). For the first time in such a cohort, repeat motifs and accurate repeat lengths of RFC1 were determined in parallel and together with repeat expansions in C9orf72 and AR by an ONT long-read sequencing method. Pathogenic biallelic repeat expansions in RFC1 in absence of another causative genetic alteration were identified in 3% of our patients, including the patient originally identified and reported as a case report [15]. Assuming a heterozygous carrier frequency of the pathogenic ‘AAGGG’ repeat motif of up to 4% in the European population resulting in an estimated prevalence of RFC1 spectrum disorder of up to 1/2500, individuals with a biallelic pathogenic repeat expansion were enriched by the factor 70 in this cohort, making a coincidence highly unlikely [1, 2, 31].
Interestingly, a recent study by Abramzon et al. investigated whether biallelic pathogenic repeat expansions in RFC1 can be detected in ALS patients. When screening a cohort of 1069 patients with a clinical diagnosis of sporadic ALS, in contrast to our study, no patient with biallelic pathogenic repeat expansions in RFC1 was found [6]. A possible explanation for these discrepant results could be the composition of the cohorts and different methods used for analyzing the RFC1 repeat array: The study of Abramzon et al. only included ALS patients from a study registry fulfilling the El Escorial criteria, whereas in this study patients with a clinical diagnosis of MND (primarily ALS) from a real-world clinical outpatient cohort were analyzed. Another possible explanation for the identification of two patients with a clinical diagnosis of ALS in this study could be the known geographic differences in the frequencies of known ALS-associated genetic variants, even across populations with European ancestry [32]. In addition, the applied diagnostic approaches differed significantly. Abramzon et al. used an iterative PCR-based workflow without accurately determining the repeat configuration of both alleles employing ONT long-read sequencing. This might have missed patients with an RFC1 spectrum disorder especially given the limited knowledge about the pathogenicity of individual motifs or a potential pathogenic SNV in individuals heterozygous for a pathogenic repeat expansion at that time [3, 33].
Initially, biallelic pathogenic repeat expansions in RFC1 were identified in patients with CANVAS, a characteristic clinical triad of cerebellar ataxia, sensory neuropathy and vestibular areflexia [1, 2]. Meanwhile, incomplete CANVAS phenotypes and a variety of additional, atypical symptoms such as autonomic dysfunction, bradykinesia, parkinsonism or dystonia expanded the phenotype of patients carrying pathogenic repeat expansions, which led to the terminology of RFC1 spectrum disorder [9, 34, 35]. The severity of the individual clinical presentation varies widely, and in some patients, might even fully resemble another disease phenotype, such as multiple system atrophy of the cerebellar type or parkinsonism [12, 36].
Recently, motor neuron affection has been reported as frequent additional symptom of RFC1 spectrum disorder [13, 14]. As such, one study identified predominant affection of either the upper motor neuron in 29% or the lower motor neuron in 18% of RFC1 patients and simultaneous affection of both, the upper and the lower motor neuron, in 16% of RFC1 patients [13]. Furthermore, as a direct morphological correlate for clinical signs of motor neuron affection, post-mortem neuropathological examination of one patient with RFC1 spectrum disorder showed moderate but significantly increased motor neuron loss, particularly in the anterior horn of the thoracic spinal cord and brainstem motor nuclei such as the hypoglossal nucleus. A potential mechanism of motor neuron affection is axonal swelling between the upper and lower motor neuron, which was observed in one RFC1 patient thus leading to synaptic dysfunction [13]. This pathomechanism seems to be different to that usually observed in patients with classical ALS, which is characterized by cytoplasmic inclusions of abnormally aggregated and posttranslationally modified tar DNA binding protein (TDP-43) in neuronal cells. Postmortem neuropathological examinations of two individuals with RFC1 spectrum disorder and motor neuron affection have shown the absence of such inclusions [35]. Thus, it seems to be the case that different pathomechanisms besides classical intraneuronal TDP-43 pathology could lead to motor neuron loss manifesting with similar clinical phenotypes in RFC1-pathology, but obviously with a slower progression rate as compared to classical ALS.
Besides being the origin of the distinct phenotype of CANVAS, biallelic pathogenic repeat expansions in RFC1 might additionally induce neurodegeneration of other functional systems via yet unknown pathways. This hypothesis is in line with the recent discussion of clinical and pathological pleiotropy of neurodegenerative diseases, where similar genetic alterations can cause different phenotypes and vice versa [37]. Independent of biallelic ‘AAGGG’ repeat expansions that cause a neurodegenerative phenotype such as CANVAS with high penetrance, other RFC1 repeat configurations could be potential genetic risk factors for a neurodegeneration such as ALS as it is known for certain intermediate and pathogenic repeat expansions such as in ATXN1, ATXN2 and HTT [37,38,39]. Furthermore, biallelic pathogenic RFC1 expansion might explain previous reports and studies with growing evidence on sensory and cerebellar involvement in ALS [40,41,42].
We statistically evaluated the configuration of the RFC1 repeat array in MND patients to study its variability and a potential enrichment of distinct patterns in this cohort. However, the identification or exclusion of specific repeat configurations as risk factor for MND cannot be performed in this study. This is due to the rather small impact of polygenic risk factors on the overall disease risk, which requires larger cohorts of MND patients as well as a cohort of healthy individuals characterized by long-read sequencing to be detected.
Instead, our data highlight the intra- and interallelic heterogeneity of the RFC1 repeat array with 6% of the alleles showing complex repeat patterns, that cannot be detected by standard PCR-based repeat analysis. Using ONT long-read sequencing for the analysis, we were able to identify what we believe to be a novel repeat motif (ACAAG)exp with a size of 180 RU. Recently, it has been hypothesized that the combination of GC-content and repeat size of a certain repeat configuration determines its pathogenicity [3]. As the GC-content in’ACAAG’ is lower compared to known pathogenic repeat motifs and the detected repeat length is rather short, we postulate a non-pathogenicity of the found repeat configuration. In summary, we present repeat expansions in RFC1 in about 3% of individuals with a clinical main phenotype of MND (i.e., two cases with ALS according to existing Gold Coast Criteria and one patient with PLS, respectively upper motor neuron dominant ALS), emphasizing that a pure or predominant motor neuron disease might be another extreme phenotype of RFC1 spectrum disorder (see Suppl. Figure 3) and thus should be considered in the genetic diagnostic workup in patients diagnosed with MND, especially when other additional symptoms like sensory and/or autonomic neuropathy, vestibular failure, cerebellar deficits and/or a chronic cough occur. For an in-depth analysis of the heterogeneous RFC1 locus including an accurate determination of its repeat size and motif, it is crucial to employ long-read sequencing, offering an unbiased approach to adequately capture and understand the complex genetics of RFC1 spectrum disorder. However, further studies are needed to validate our findings and to definitely assess the role of pathogenic motifs and repeats in RFC1 for the upper and lower motor neurons as well as to analyze a possible pathogenic role of novel repeat motifs with regard to a polygenic risk.






留言 (0)