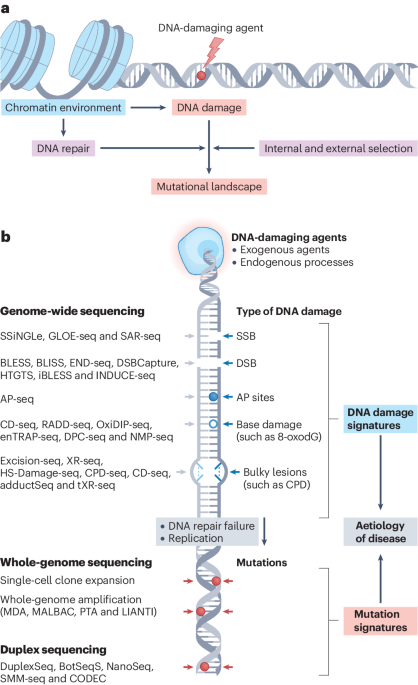
Tandem repeats are ubiquitous in the human genome and hold crucial information about our genetic diversity, evolution and susceptibility to disease.
Tandem repeats are repetitive sequences that vary in length — from a few base pairs for short tandem repeats (STRs) or hundreds of base pairs in variable number tandem repeats (VNTRs) to thousands of base pairs for the satellite DNA found at centromeres and telomeres. Our July 2024 issue showcases how recent technological and computational advances are paving the way to a more comprehensive view of these repetitive regions in our genomes.
Tandem repeats are a prominent source of genetic variation that has been difficult to capture accurately at the population level using short-read sequencing methods. Their repetitive nature, combined with the short read length, often lead to sequencing, alignment, assembly or amplification errors, making it challenging to determine the exact sequence composition and number of repeat units within each repetitive region. In their Review, Tanudisastro et al.1 discuss recent improvements in short-read genotyping approaches, as well as how long-read sequencing platforms have overcome existing limitations by generating reads that span entire STR alleles. These breakthroughs in identifying and genotyping STRs are enabling the exploration of this type of genetic variation at the population scale for a range of applications.
STRs have a pivotal role in contributing to the diversity within and between populations, as illustrated in a Journal Club by Ning Xie2. By analysing the distribution of specific STR alleles among different populations, researchers can identify relationships between different groups or trace ancient migration routes, providing insights into the evolutionary histories of human populations.
The inherent variability in the number of repeats between individuals makes STRs an invaluable tool not only in population genetics but also in forensics and paternity testing, as they form a ‘genetic fingerprint’ for each individual. In a Comment, Budowle and Sajantila3 recount how forensic science witnessed a revolution with the advent of STR analysis, which had a considerable role in enhancing the accuracy and reliability of DNA profiling. The unique STR profiles enable forensic scientists to match crime scene samples to potential suspects or identify missing persons, providing a powerful tool in the pursuit of justice.
Long-read sequencing offers unparalleled accuracy in identifying the number and sequence of repeats, including large pathogenic repeat expansions. Disruptions in repetitive sequences have been linked to a myriad of genetic disorders, ranging from neurological disorders to cancer susceptibility. For example, Huntington disease and fragile X syndrome are directly linked to the expansion of STRs within a specific gene. The clinical phenotype of these repeat expansion disorders differs based on the length of the STR region and the composition of its sequence, including the presence of non-canonical motifs or repeat interruptions. A Review by Rajan-Babu et al.4 discusses how to leverage molecular assays and sequencing-based technologies to detect repeat interruptions and non-reference expansions. They also review the clinical effects of altered sequence composition and underlying disease mechanisms.
Beyond monogenic disorders, population-scale studies are beginning to reveal a causal role for tandem repeat variation in complex traits, as emphasized in a Comment by Lamkin and Gymrek5. Expanding existing studies to disease phenotypes, considering more tandem repeats across the genome, and accounting for non-linear associations promises to increase the number of tandem repeats implicated as causal variants.
Finally, a Research Highlight6 summarizes a recent study that used long-read sequencing to obtain the complete sequence for the α-satellite DNA of all centromeres in a single human genome, enabling comparative analyses of this rapidly evolving region with existing human and non-human primate data.
“Recent advances … have empowered researchers to detect and characterize tandem repeats with unparalleled precision”
Recent advances, including long-read sequencing and improved computational approaches, have empowered researchers to detect and characterize tandem repeats with unparalleled precision, driving the exploration of one of the largest sources of genetic variation in humans with substantial effects on health and disease.



留言 (0)