
記住我
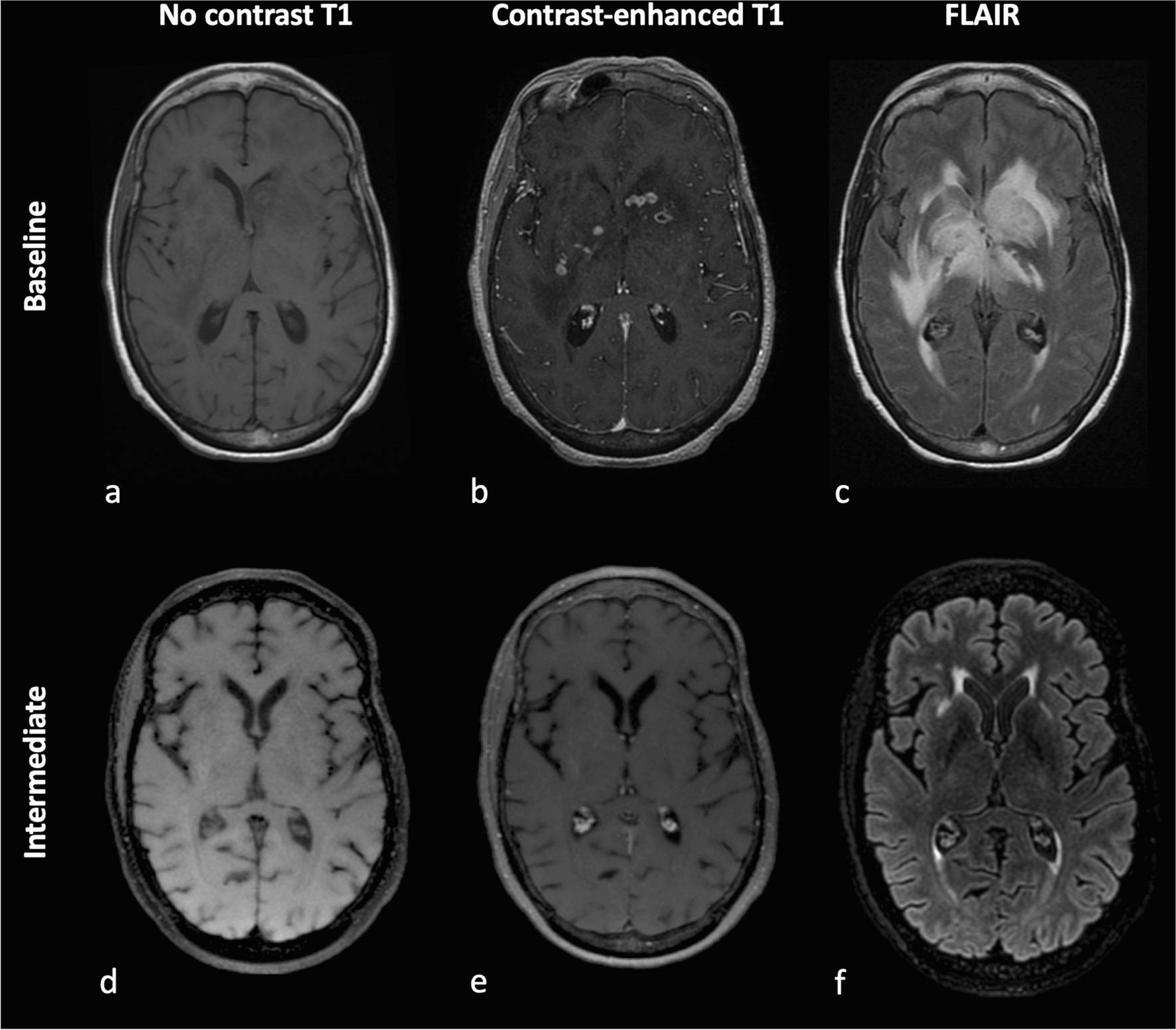
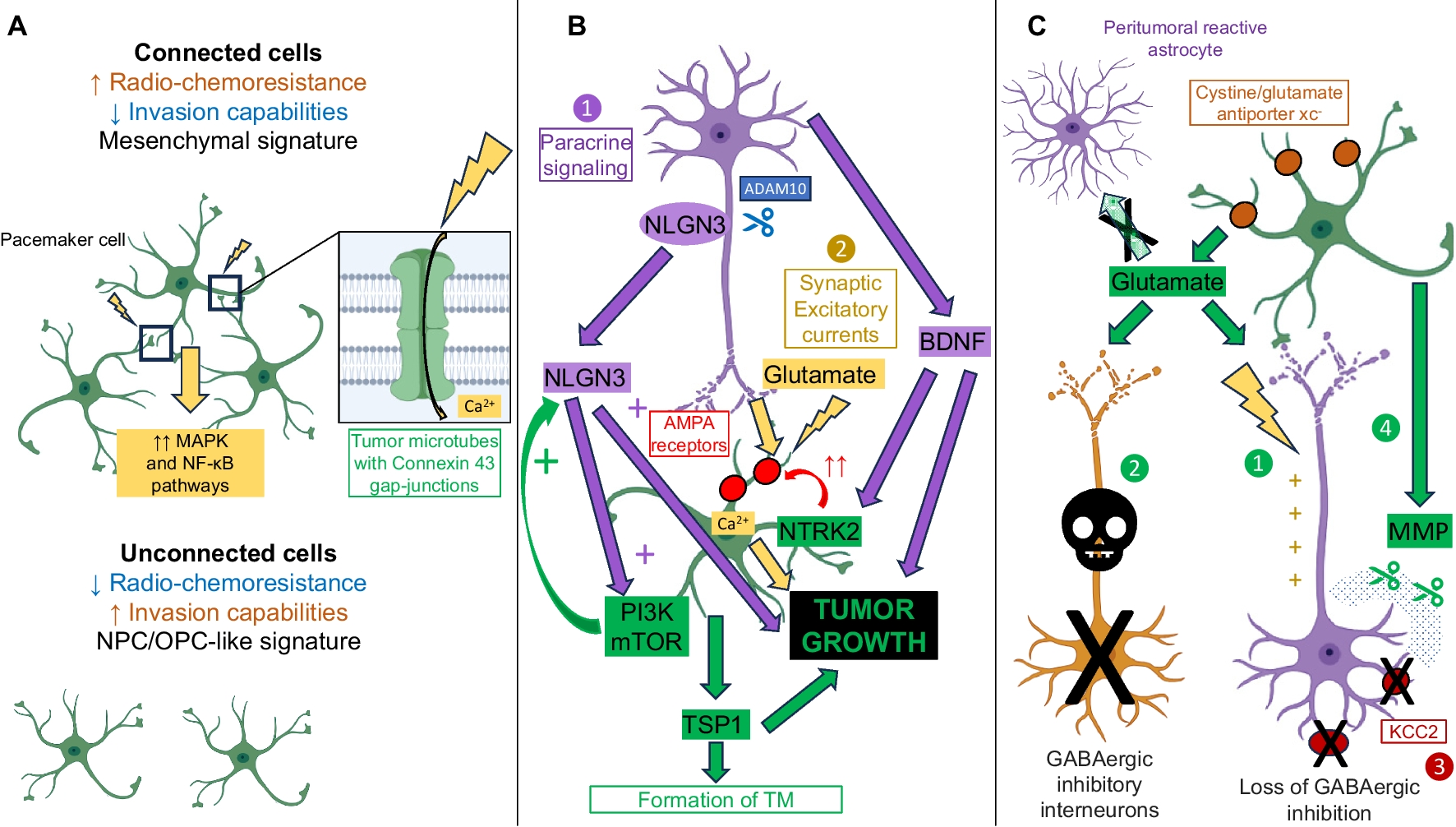
In vitro and in vivo models offer the opportunity to understand the complex bidirectional interactions between neurons and malignant glial cells, detailed in Fig. 1, within the tumour microenvironment, at the cellular and molecular levels (Table 1).
Fig. 1
Simplified representation of the interactions between tumours cells and the bidirectional crosstalk between neurons and tumour cells (created with Biorender). A. Interactions between tumour cells. Tumor cells (green) form an extensive network, mediated by tumor microtubes that bear Connexin 43 gap-junctions, and are resistant to radio-chemotherapy. Within the network, pacemaker-like cells, thanks to KCa 3.1 channels (yellow), display rhythmic Ca2+ oscillations that are transmitted to the network and that drive tumour aggressiveness via activation of the MAPK and NF-κB pathways. Unconnected cells have increased invasion abilities. B. Neuronal activity-regulated tumour growth. First, neurons (purple) emit a paracrine signaling, notably mediated by BDNF and NLGN3, that stimulates tumour growth. NLGN3 is clived from neural cells by the ADAM10 sheddase. NLGN3 activates the PI3K-mTOR pathway and feedforwards its expression. Second, through excitatory glutamate neuro-gliomal synapses, whose establishment is eased by NLGN3, neurons activity stimulates tumors growth. BDNF binding to NTRK2 receptors increases the expression of AMPA receptors (red), which strengthen glutamate signaling. The neuron-dependent secretion of TSP1 by tumour cells contributes to tumour microtube formation and glioma progression. C. Glioma-induced neuronal activity modifications. Glutamate is released by glioma cells through the cystine/glutamate antiporter xc− (brown). Peritumoral reactive astrocytes have a decreased capability to uptake glutamate. First, the increased glutamate rate in the tumour microenvironment induces neuronal hyperexcitability. Second, glutamate-toxicity leads to the death of fast-spiking GABAergic inhibitory neurons. Third, the drop in the neuronal expression of the potassium/chloride transporter KCC2 (dark red) is responsible for the switch from inhibitory to excitatory of GABA signaling. Fourth, the disruption of perineuronal nets by glioma-secreted matrix metalloproteinases amplifies the loss of GABAergic inhibition and glutamate-induced neuronal death
Table 1 Principal techniques and neuroscience tools used to study glioma-induced malignant circuit remodeling at the microscopic levelGlioma-glioma networks promote glioma growth and treatment resistanceMalignant astrocytoma mouse xenografted tumors analyzed with in vivo multiphoton laser-scanning microscopy uncovered the presence of Tumour Microtubes (TM), that correspond to ultra-long membrane protrusions, stabilized by p120 catenin. Through TM, glioma cells form an extensive and non-randomly organized network mediated by Connexin 43 gap junctions [21] and exchange ions and molecules. The neuronal growth-associated protein GAP-43 conditions TM formation and function and, in concert with the membrane protein linked to neuronal development TTYH1, drives TM-mediated invasion properties and proliferation [5, 6, 22, 23]. TM also establish connections between tumor cells and non-tumoral astrocytes [7]. NOTCH1 regulates network connectivity and its downregulation induces TM extension [8]. A subpopulation of highly connected pacemaker-like glioma cells display rhythmic Ca2+ oscillations, relying on the calcium-dependent potassic channel KCa3.1. These Ca2+ oscillations are transmitted to the network and activate the frequency-dependent MAPK and NF-κB pathways [6, 24]. TM are involved in treatment failures. First, they ease repopulation of the surgical cavity after glioma resection [9]. Second, connected cells are less susceptible to radiation-induced [5] and chemotherapy-induced cytotoxicity than unconnected cells [9], but have however decreased invasion abilities [5, 7, 8, 23]. Connected and unconnected cells express a mesenchymal-like and a neural/oligodendroglial precursor-like signature, respectively [7].
Pharmacological inhibition of Connexin 43 or gap junction but also meclofenamate inhibition of intercellular cytosolic traffic via gap junctions reduced glioblastoma cell resistance to Temozolomide and Lomustine, independently of MGMT status [25,26,27]. Consistently, the genetic inactivation of Connexin 43 or GAP-43 and the senicapoc pharmacological inhibitor of potassic channel KCa3.1 slowed down xenograft progression in mice [5, 6, 9]. TTYH1 knockdown decreased the rate of invasive tumour cells harboring one or two TM but not the rate of hyperconnected cells harboring more than four TM, unveiling a functional and molecular heterogeneity among TM [23]. Consequently, TM formation and function could offer new therapeutic targets.
Neuronal activity drives malignant glioma growthThrough the use of in vivo optogenetic control of cortical neuronal activity in patient-derived pediatric glioblastoma xenograft models in mice expressing the excitatory opsin channelrhodopsin-2, it was demonstrated that neurons stimulate glioma growth through paracrine signaling mediated by Brain-Derived Neurotrophic Factor (BDNF) and the soluble synaptic adhesion protein neuroligin-3 (NLGN3) [10,11,12]. NLGN3 promotes tumour growth and feedforwards its own expression in glioma cells, through induction of the PI3K-mTOR pathway activity. Accordingly, NLGN3 expression is inversely correlated with overall survival [11] and conditions the growth of many subtypes of pediatric and adult gliomas [12]. NLGN3 is cleaved from neurons and oligodendrocytes precursor cells by the ADAM10 shedddase. ADAM10 inhibition reduces xenograft growth by preventing NLGN3 release into the tumour microenvironment and could represent the basis of new therapeutic strategies [12]. The relevance of INCB7839, an ADAM 10 and 17 inhibitor, for the treatment of pediatric high-grade gliomas is assessed in an ongoing randomized clinical trial (NTC04295759).
NLGN3 paracrine signaling upregulates the expression of several synapse-related genes [12], and promotes the establishment of aberrant synapses, localized on TM. The electrochemical communications between presynaptic neurons and postsynaptic malignant glial cells is mediated by glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs) [10, 14]. AMPAR trafficking to the glioma cell membrane is promoted by docking of BDNF to the receptor NTRK2, resulting in an increased amplitude of glutamate-evoked currents in the malignant cells. Pharmacological or genetical inhibition of NTRK2 decreases synaptogenic mechanisms and prolongs survival in xenograft models [15]. A high-neural epigenetic signature associated to overexpression of synaptic genes was consistently correlated with increased connectivity according to functional Magnetic Resonance Imaging (fMRI) and Magneto-Electro-Encephalography (MEG) [28]. Additionally, the tumor cell subpopulation which overexpresses synaptic genes is mainly of oligodendroglial or neural progenitor-like signature [10, 28, 29] but the link between synaptic transmission and stemness phenotype still remains to be elucidated.
Neuronal activity evokes excitatory post-synaptic currents but also non-synaptic activity-dependent currents, respectively calcium- and potassium-mediated, that are amplified by gap junction-mediated tumour interconnections through TM, forming an electrically coupled network [10, 14]. Synaptic and electrical integration of glioma into neural circuits, and glutamate binding to AMPARs favor tumour progression. Indeed, neuronal activity drives TM dynamics and increases the number of TM branching events via calcium signaling, but also the invasion speed of unconnected glioma cells via transient neuro-gliomal synapses with AMPARs [7, 14, 30]. Moreover, depolarization of glioma cell membranes promotes proliferation [10]. Consistently, the occurrence of gliomas is higher in brain regions which display higher intrinsic activity levels according to MEG [31]. Furthermore, high-functionally connected tumour regions are enriched in a tumor cell subpopulation with synaptogenic properties, which develops TM and proliferates on the presence of neurons, under the dependence of glioma-secreted Thrombospondin 1 (TSP1). Interestingly, the presence of high-functional connectivity areas within tumour negatively influence cognitive and survival prognosis [13]. However, remote activity-dependent glioma progression mechanisms were recently identified. They are driven by an infiltrating cell population highly-expressing axon guidance genes, notably SEMA4F, and whose activity depends on contralateral callosal projection neurons [32].
Cytostatic effects are observed following pharmacological electrochemical signaling blockade [33], using the AMPAR-blocking drug perampanel, meclofenamate [10, 14] or using gabapentin which both inhibit TSP1 and the branched-chain amino acid aminotransferase (BCAT1) thus lowering extracellular glutamate level [13, 34]. The anti-tumoral effects of glutamate signaling inhibitors, including gabapentin, meclofenamate and perampanel, are assessed in several on-going trials [16, 35, 36].
Malignant glioma proliferation promotes neuronal hyperexcitabilityElectroencephalograms performed in glioma murine models demonstrate spontaneous and recurring abnormal events, compatible with progressive epileptic activity, with a concurrent enrichment in a tumor cell population expressing synaptic genes, suggesting propagation of synapse associated genes [17]. Cortical slices following malignant glioma implantation demonstrate increased glutamate release from the tumor mass, mediated by the cystine/glutamate antiporter xc− [17, 18, 30], whose expression is anticorrelated to this of p53 [19]. Moreover, peritumoral reactive astrocytes have a decreased ability to uptake glutamate and potassium. Some peritumoral astrocytes also display a depolarized resting membrane potential, further contributing to alter potassium and glutamate homeostasis [20]. A resultant glutamatergic epileptiform hyperexcitability spreading into peritumoral areas was identified by extracellular field recordings at sites distant from glioma [17], consistently with the results of human intraoperative ECoG recordings [10]. Extracellular field recordings performed on brain slices taken from xenograft models revealed that the onset latency of magnesium-free-induced epileptiform activity was shorter than in healthy slices. Moreover, the incidence of ictal-like events was higher. Blockade of the cystine/glutamate antiporter xc− and thus glutamate release from the tumour mass using sulfasalazine decreased hyperexcitability and was able to reduce not only the frequency of epileptic events in tumour-bearing mice [17] but also neuronal activity-regulated glioma growth [37].
Independently, peritumoral cortex taken from mice orthotopic xenografts also displays a distance-dependent loss of parvalbumin-positive fast-spiking GABAergic inhibitory interneurons, attributable to glutamate neurotoxicity, and a reduced neuronal expression of the potassium/chloride plasmalemmal transporter KCC2, changing response to GABA from inhibitory to excitatory potentials [33, 38, 39]. Additionally, perineuronal nets, which represent a complex lattice-like extracellular matrix, acting as an electrostatic insulator that reduces specific membrane capacitance, are degraded in a distance-dependent manner by glioma-released matrix metalloproteinases. This mechanism amplifies the loss of GABAergic inhibition but also glutamate-induced neuronal death [39].
Taken together, these results demonstrate that hyperexcitability results from peritumoral synaptic network disruption in the setting of malignant gliomas [20, 37, 38] and suggest new therapeutic targets for controlling peritumoral hyperexcitability. Interestingly, in a mouse model, it was evidenced that some hotspot-mutations of PI3KCA induced increased network hyperexcitability compared to others. Notably, neurons adjacent to C420R- and H1047R-mutant gliomas displayed enhanced synaptic imbalance, which was attributed to the secretion of glypican 3 in the case of C420R variant [40], bridging genetics with peritumoral synaptic remodeling.
Network level evidence of nervous system regulation of glioma proliferation and invasionResting state and task-based investigations are available to indirectly and macroscopically probe functional plasticity mechanisms and circuit remodeling caused by malignant gliomas (detailed in Fig. 2 and Table 2). Network level understanding of glioma development has recently come into clearer view because of emerging technologies.
Fig. 2
Representation of the different tools offering the possibility to study glioma-induced malignant circuit remodeling as well as nervous system regulation of glioma proliferation and invasion at the macroscopic level (created with Biorender)
Table 2 Principles and characteristics of the different techniques dedicated to study glioma-induced malignant circuit remodeling at the macroscopic levelFunctional MRI (fMRI)Four different glioma-induced remodeling patterns in patients have been identified using fMRI with the appearance of additional activation sites within (1) the tumor, (2) the ipsilateral peritumoral cortex, (3) the distant ipsilateral cortex, and (4) the contralateral normal appearing cortex [41]. Indeed, in a series of patients with gliomas located at an average distance of 3.9 ± 3.5 cm from the hand motor region, 62% and 46% of patients exhibited an ipsilateral and contralateral recruitment, respectively. Ipsilateral recruitment decreased as tumour volume increased and distance from primary motor cortex decreased and vice versa [42]. However, patients with gliomas located in or near motor areas, but without any motor deficit, had a significant reduction in inter-hemispheric functional connectivity between bilateral primary motor cortices, compared to age-matched healthy controls [43]. Regarding the language network, right-handed patients with a left hemisphere glioma also had a global reduction of bilateral functional connectivity, compared to healthy controls. The most affected node was the left temporo-parietal junction [44].
According to resting-state fMRI, compared to healthy controls, patients with glioma had a decreased functional connectivity concerning the whole-brain, and not restricted to the lesional hemisphere [45, 46]. The importance of these alterations was correlated with high tumor grade, negative IDH status and decreased neuropsychological performances but not with tumour size or location [45]. Besides, Default Mode Network connectivity was modified in patients managed for left-sided gliomas, with increased and decreased integration in hippocampal and prefrontal areas, respectively [47]. Finally, a higher intra-network functional connectivity strength within glioblastoma was found to be independent of tumour size but predicted a better overall survival [48].
Positron Emission Tomography (PET)One of the first studies demonstrating remodeled network connectivity caused by chronic disease of the central nervous system was identified using PET imaging of amyloid beta in patients with Alzheimer’s disease [49]. PET of gliomas located in the hand motor region showed that, compared to the unaffected side, the activations were shifted by 20 ± 13 mm (SD), either along the mediolateral body representation of motor cortex or into premotor or parietal somatosensory cortex [50]. Additional activation of the supplementary motor area was occasionally present [50]. Regarding the plasticity of the language network, two compensatory mechanisms, whose occurrence depended on tumour location, were identified. First, at the intra-hemispheric level, left fronto-lateral regions other than classical language areas can be recruited. Second, at the inter-hemispheric level, fronto-lateral activation can appear in the right nondominant hemisphere, especially in patients with frontal or temporal posterior tumours, possibly representing a loss of transcallosal collateral inhibition [51].
MagnetoEncephaloGraphy (MEG)The comparison of whole-brain activation motor maps performed in the same patients at initial diagnosis and compared with first recurrence has demonstrated a shift in activation peaks in the ipsilateral and contralateral motor cortices [52]. Specifically, motor activity following glioma progression is associated with contralesional hemisphere activation for speech and motor tasks. Tumor location, presence of a motor impairment and longer time lapse were associated with greater cortical remodeling [
留言 (0)